A Method to Isolate Pericytes From the Mouse Urinary Bladder for the Study of Diabetic Bladder Dysfunction
Article information

Abstract
Purpose
Pericytes surround the endothelial cells in microvessels and play a distinct role in controlling vascular permeability and maturation. The loss of pericyte function is known to be associated with diabetic retinopathy and erectile dysfunction. This study aimed to establish a technique for the isolation of pericytes from the mouse urinary bladder and an in vitro model that mimics in vivo diabetic bladder dysfunction.
Methods
To avoid contamination with epithelial cells, the urothelial layer was meticulously removed from the underlying submucosa and detrusor muscle layer. The tissues were cut into multiple pieces, and the fragmented tissues were settled by gravity into collagen I-coated culture plates. The cells were cultured under normal-glucose (5 mmol/L) or high-glucose (30 mmol/L) conditions, and tube formation, cell proliferation, and TUNEL assays were performed. We also performed hydroethidine staining to measure superoxide anion production.
Results
We successfully isolated high-purity pericytes from the mouse urinary bladder. The cells were positively stained for platelet-derived growth factor receptor-β and NG2 and negatively stained for smooth muscle cell markers (desmin and myosin) and an endothelial cell marker (CD31). The number of tubes formed and the number of proliferating cells were significantly lower when the pericytes were exposed to high-glucose conditions compared with normal-glucose conditions. In addition, there were significant increases in superoxide anion production and the number of apoptotic cells when the pericytes were cultured under high-glucose conditions.
Conclusions
To the best of our knowledge, this is the first study to isolate and culture pericytes from the mouse urinary bladder. Our model would be a useful tool for screening the efficacy of therapeutic candidates targeting pericyte function in diabetic bladder dysfunction and exploring the functional role of specific targets at the cellular level.
• HIGHLIGHTS
- We established a protocol to isolate pericytes from the mouse urinary bladder and created an in vitro model of diabetic pericyte dysfunction.
- This model would be a useful tool to understand the role of pericytes in the pathogenesis of diabetic bladder dysfunction.
INTRODUCTION
The urinary bladder is an organ highly enriched with blood vessels and nerve fibers [1-4]. Recently, accumulating evidence has identified bladder ischemia as a common pathophysiologic denominator for lower urinary tract symptoms (LUTS) [5]. A number of vascular risk factors such as hypertension, dyslipidemia, and diabetes mellitus, and cardiovascular diseases are known to be associated with LUTS. The severity of symptoms based on the International Prostate Symptom Score is correlated with the number of vascular risk factors [6,7].
Diabetic bladder dysfunction (DBD) includes a broad spectrum of LUTS, including an overactive bladder in the early stage of diabetes and an underactive bladder characterized by decreased urinary sensation and flow rate, increased bladder capacity, and incomplete bladder emptying in the late stage of the disease [8]. The prevalence of DBD ranges between 25% and 90% [9]. Polyuria, microvascular injury, and peripheral neuropathy are the major pathophysiologic factors contributing to this condition [8]. In addition, the increased production of reactive oxygen species (ROS) from hyperglycemia has detrimental effects on the detrusor smooth muscle (i.e., diminished contractile response to a variety of stimulations) and nervous system [10,11].
The urothelium can provide an efficient permeability barrier owing to the unique properties of umbrella cells including the glycosaminoglycan layer on the cell surface and well-developed tight junctions [12,13]. The microvasculature plays a fundamental role in exchanging substances between the circulatory system and tissues. Adequate blood supply to the mucosal layer of the urinary bladder is necessary for maintaining its storage and permeability functions as well as ensuring a nutrient supply [14]. Similar to diabetic retinopathy and nephropathy, diabetic microangiopathy may also impair the innervation of the urinary bladder and cause the dysfunction of the detrusor muscle and urothelium [8].
Pericytes cover the surface of microvascular endothelial cells and play a distinct role in controlling vascular permeability and maturation in various organs [15]. The functional and structural derangements of pericytes are a key feature of diabetic retinopathy [16] and erectile dysfunction [17]. We recently reported the distinct distribution of pericytes in the mouse urinary bladder [14]. In addition, the loss of pericytes in erectile tissues and the resulting increase in the leakiness of cavernous sinusoids were observed in a mouse model of diabetic erectile dysfunction [17].
Several studies have described the role of diabetes in the pathogenesis of diabetic complications in the smooth muscle and nervous system of the urinary bladder [8,10,11]; however, the direct effect of hyperglycemia or diabetes on the vascular pericytes of the urinary bladder has not been investigated in detail. Moreover, to the best of our knowledge, there are no reports about the primary isolation and culture of pericytes from the urinary bladder. Although diabetic animal models provide crucial information to understand the pathophysiology of DBD, these models cannot address the complex causes of DBD at cellular and molecular levels. Moreover, although pericyte cell lines from the brain and retina are commercially available [18,19], they do not accurately represent the characteristics of urinary bladder pericytes. Therefore, it is necessary to isolate pericytes from the urinary bladder to clarify organ-specific pathobiology.
In the present study, we established a technique to isolate pericytes from the mouse urinary bladder. To mimic the in vivo conditions of DBD, urinary bladder pericytes were cultured under normal-glucose or high-glucose conditions, and the effects of different glucose concentrations on pericyte proliferation, apoptosis, and tube formation were determined.
MATERIALS AND METHODS
Preparation of Mouse Urinary Bladder
Specific pathogen-free C57BL/6 mice were purchased from Orient Bio (Seongnam, Korea). The experimental protocol was approved by the Institutional Animal Care and Use Subcommittee of Inha University (approval number: INHA 190813-661). Ten-week-old male mice were used in this study. Mouse urinary bladder tissues were obtained according to protocol, and all procedures were performed with sterile techniques. The urinary bladder was harvested and washed with Hank’s balanced salt solution (GIBCO, Carlsbad, CA, USA) in 100-mm disposable Petri dishes. The adjacent connective and adipose tissues were removed. The urinary bladder was opened longitudinally, and the urothelial layer was removed by gentle swabbing with a Q-tip to avoid contamination with epithelial cells as described previously [14] (Fig. 1A). All procedures were performed with the aid of a dissecting microscope (Zeiss, Göttingen, Germany). The remaining urinary bladder tissues were washed several times with phosphate-buffered saline (PBS; GIBCO) and used for primary pericyte culture.

Schematic illustration of the isolation of pericytes from the mouse urinary bladder. (A) The urinary bladder was opened longitudinally, and the urothelial layer was removed. The remaining urinary bladder tissues were cut into multiple pieces (2–3 mm) and implanted on collagen I-coated dishes containing pericyte culture medium. (B) Phase-contrast images of primary cultured cells. The cells showed typical pericyte characteristics with polymorphic and multidirectional projections. (C) Phase-contrast images showing contamination with epithelial cells.
Isolation and Culture of Pericytes From Mouse Urinary Bladder
Mouse urinary bladder pericytes were prepared and maintained as previously described for erectile tissues [17,20]. In brief, urinary bladder tissues were cut into several pieces (2–3 mm), and the tiny tissue fragments were settled by gravity into collagen Icoated 35-mm cell culture dishes (Becton Dickinson, Mountain View, CA, USA) (Fig. 1A). After the dishes were incubated for 30 minutes at 37°C with 300 μL of pericyte-specific growth medium, 900 μL of pericyte culture medium was further added, and the samples were incubated at 37°C with 5% CO2. The pericyte growth medium consisted of 20% fetal bovine serum, 1% penicillin/streptomycin, 1 × pericyte growth supplement (ScienCell, Carlsbad, CA, USA), 10nM human pigment epithelium-derived factor (Sigma-Aldrich, St. Louis, MO, USA), and low-glucose Dulbecco’s modified Eagle medium (GIBCO). The medium was changed every 2 days until the cells were confluent and spread over the bottom of the dish (~2 weeks after the start of culture). Only sprouting cells were used for subculture (Fig. 1B).
Characterization of Isolated Cells
The cells were cultured for 48 hours on sterile cover glasses in 12-well plates and fixed in 4% paraformaldehyde for 15 minutes at 4°C. The cells were then blocked with Antibody Dilution Buffer (Invitrogen, Camarillo, CA, USA) for 1 hour at room temperature. The isolated cells were characterized with antibodies against platelet-derived growth factor receptor-β (PDGFR-β, a pericyte marker, 1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), NG2 chondroitin sulfate proteoglycan (a pericyte marker, 1:500; Millipore, San Francisco, CA, USA), and angiopoietin-1 (Ang1, an angiogenic growth factor mainly secreted from pericytes, 1:50; Santa Cruz Biotechnology) as positive markers for pericytes and stained with antibodies against desmin (a smooth muscle cell marker, 1:500; Abcam, Cambridge, MA, USA), myosin (a smooth muscle cell marker, 1:500; Sigma-Aldrich), CD34 (an endothelial cell marker, 1:500; Santa Cruz Biotechnology), CD-31 (an endothelial cell marker, 1:500; Millipore), CD90 (a fibroblast marker, 1:500; R&D Systems, Minneapolis, MN, USA), and E-cadherin (an epithelial cell marker, 1:500; Abcam) as negative markers. After washing twice with PBS, the cells were incubated with antifluorescein or rhodamine-labeled secondary antibody (1:500; Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA) for 2 hours at room temperature. The cells were mounted in a solution containing 4,6-diamidino-2-phenylindole (DAPI; Vector Laboratories Inc., Burlingame, CA, USA) for nuclei staining. Human placental pericytes were used as a positive control, and A7r5 smooth muscle cells were used as a negative control. Signals were visualized, and digital images were obtained with a confocal microscope (FV1000; Olympus, Tokyo, Japan).
In Vitro Tube Formation Assay
To evaluate the effect of glucose concentrations on the angiogenic potency of primary cultured pericytes, the cells were cultured under normal-glucose (5 mmol/L) or high-glucose (30 mmol/L) conditions for 48 hours as shown in previous studies [21,22]. Tube formation assay was performed as previously described [17,20]. The pericytes were seeded onto Growth FactorReduced (GFR) Matrigel (Becton Dickinson) at 5×104 cells/well, and the plates were incubated at 37°C for 6 hours in a CO2 incubator. Tube formation was observed under a phase-contrast microscope, and the number of master junctions was measured using National Institutes of Health (NIH) ImageJ ver. 1.50 (Bethesda, MD, USA).
TUNEL Assay
Pericytes were cultured under normal-glucose (5 mmol/L) or high-glucose (30 mmol/L) conditions for 48 hours on sterile coverslips in 12-well plates. The cells were fixed in 4% paraformaldehyde for 15 minutes at 4°C and further fixed in precooled methanol for 10 minutes at -20°C. The fixed cells were washed twice with PBS for 5 minutes. TUNEL assay was performed using the ApopTag Plus Fluorescein In Situ Apoptosis Detection Kit (Chemicon, Temecula, CA, USA). Samples were mounted in a solution containing DAPI (Vector Laboratories Inc.) for nuclei staining. The result was quantified using NIH ImageJ software.
BrdU Proliferation Assay
The pericytes were cultured under normal-glucose (5 mmol/L) or high-glucose (30 mmol/L) conditions for 48 hours on sterile coverslips in 12-well plates and incubated for another 24 hours in medium containing BrdU (Invitrogen). The cells were washed twice with PBS and fixed in an acetic acid:ethanol (2:1) solution for 10 minutes at -20°C. Following fixation, the cells were washed with PBS. The BrdU-labeled cells were treated with 1 N HCl for 1 hour at room temperature to hydrolyze the DNA structure of the BrdU-labeled cells. After denaturation, the cells were neutralized with 0.1M sodium borate buffer (pH, 8.5) for 30 minutes at room temperature and washed 3 times with PBS. The cells were blocked with Antibody Dilution Buffer (Invitrogen) for 1 hour at room temperature and incubated with anti-BrdU antibody (1:500; AbD Serotec, Kidlington, UK) overnight at 4°C. After washing twice with PBS, the cells were incubated with anti-rat fluorescein-labeled secondary antibody (1:500; Jackson ImmunoResearch Laboratories Inc.) for 2 hours at room temperature. The cells were mounted in a solution containing DAPI. The number of proliferating cells was measured using NIH ImageJ software.
In Situ Detection of Superoxide Anion
To detect the generation of intracellular superoxide anions, hydroethidine (1:5,000; Molecular Probes, Eugene, OR, USA), a cell-permeable fluorescence probe, was used. Hydroethidine (2 µmoL/L) was added to each sample, and the slides were incubated in a light‐protected humidified chamber at 37°C for 30 minutes. After washing with PBS, the cells were mounted in a solution containing DAPI. The fluorescence intensity of hydroethidine was measured using NIH ImageJ software.
Statistical Analysis
Results are expressed as the mean ±standard error. MannWhitney U-tests were used for group comparisons. Statistical analyses were performed with SigmaStat 3.5 software (Systat Software Inc., Richmond, CA, USA). P-values of less than 5% were considered significant.
RESULTS
Isolation of Pericytes from the Mouse Urinary Bladder
Representative phase-contrast images of the cells cultured for 2 weeks are shown in Fig. 1B. After the cells were confluent and spread over the bottom (~2 weeks after the start of culture), only sprouting cells were used for subculture (Fig. 1B). Similar to our previous findings for erectile tissues [20], the primary cultured cells demonstrated typical pericyte morphologic characteristics with multidirectional projections (Fig. 1B). Although we carefully removed the urothelial layer with the aid of a dissecting microscope, we sometimes encountered contamination with epithelial cells. Epithelial cells are morphologically different from pericytes and should not be used for subculture (Fig. 1C).
Characterization of Primary Cultured Cells
Primary cultured cells were positively stained for pericyte markers (PDGFR-β and NG2) but not smooth muscle cell markers (desmin and myosin), endothelial cell markers (CD31 and CD34), a fibroblast marker (CD90), and an epithelial cell marker (E-cadherin). The cells were also positively stained for Ang1, which is mainly secreted from pericytes [23] (Fig. 2A). We used human placental pericytes as a positive control and A7r5 smooth muscle cells as a negative control (Fig. 2B).

Characterization of primary cultured cells. (A) Fluorescent immunocytochemistry of primary cultured cells with antibodies against the pericyte markers platelet-derived growth factor receptor-β (PDGFR-β), NG2, and angiopoietin-1 (Ang-1); the smooth muscle cell markers desmin and myosin; the endothelial cell markers CD34 and CD31; the fibroblast marker CD90; and the epithelial cell marker E-cadherin. Nuclei were labeled with the DNA dye 4,6-diamidino-2-phenylindole (DAPI). Scale bar=100 μm. (B) Human placental pericytes were used as a positive control, and A7r5 smooth muscle cells were used as a negative control. Scale bar=100 μm. NG2, neuron‐glial antigen 2; E-cad, E-cadherin; PECAM, platelet endothelial cell adhesion molecule.
Impaired Tube Formation of Urinary Bladder Pericytes Exposed to High-glucose Conditions
Matrigel-based tube formation assay was performed to determine whether primary cultured pericytes derived from the mouse urinary bladder can form capillary-like structures and to investigate the effect of high-glucose conditions on tube formation. After 48 hours of incubation on GFR Matrigel, urinary bladder pericytes exposed to normal-glucose conditions formed nicely arranged capillary-like structures, whereas the tube formation of pericytes cultured under high-glucose conditions was severely impaired (Fig. 3).

Impaired tube formation of mouse urinary bladder pericytes exposed to high-glucose (HG) conditions. (A) After culture under normal-glucose (NG; 5 mmol/L) or HG (30 mmol/L) conditions for 48 hours, the cells (5×104 cells/well) were seeded onto the Growth Factor-Reduced Matrigel-coated culture plates. Tube formation was observed under a phase-contrast microscope, and the number of master junctions was measured using NIH ImageJ software. Magnification, ×40. Scale bar=500 μm. (B) Number of master junctions per field (screen magnification, ×40). Each bar represents the mean value (±standard error) of 6 separate wells per group. *P<0.05 compared with the NG group.
Increased Apoptosis and Decreased Cell Proliferation of Urinary Bladder Pericytes Exposed to High-Glucose Conditions
Based on TUNEL assay, the number of apoptotic cells in the urinary bladder was significantly higher when the pericytes were exposed to high-glucose conditions compared with normal-glucose conditions (Fig. 4A, B). We also assessed the effects of highglucose conditions on the proliferation of the pericytes. The number of BrdU-labeled proliferating cells was markedly decreased when the pericytes were exposed to high-glucose conditions compared with normal-glucose conditions (Fig. 4C, D).
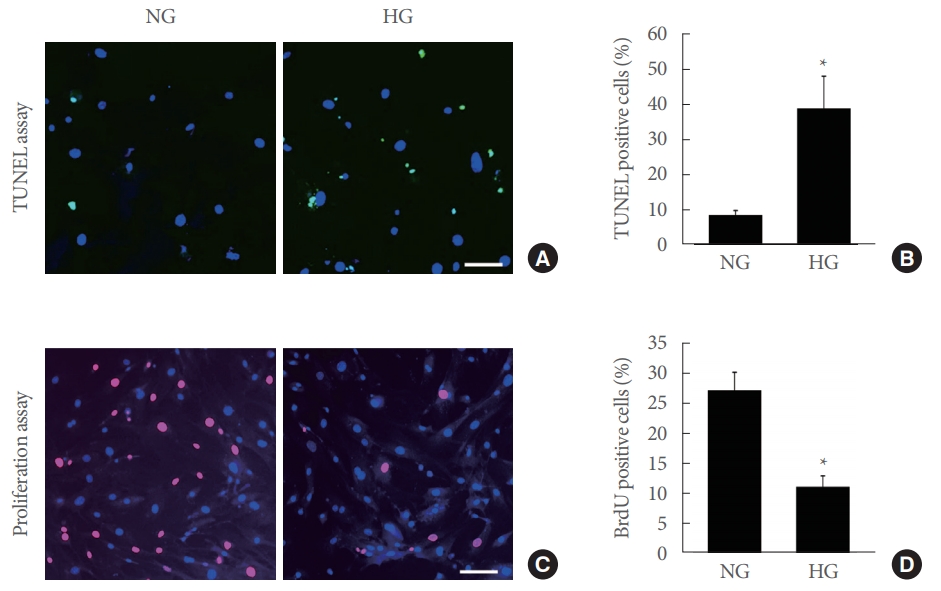
Increased apoptosis and decreased cell proliferation of mouse urinary bladder pericytes exposed to high-glucose (HG) conditions. (A) Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay. Pericytes were incubated under normal-glucose (NG; 5 mmol/L) or HG (30 mmol/L) conditions for 48 hours. Nuclei were labeled with the DNA dye 4,6-diamidino-2-phenylindole. Scale bar=100 μm. (B) Number of apoptotic cells per field. Each bar represents the mean value (±standard error) of 6 separate wells per group. *P<0.05 compared with the NG group. (C) Cell proliferation assay. Scale bar=100 μm. (D) Number of proliferating cells per field. Each bar represents the mean value (±standard error) of 6 separate wells per group. *P<0.05 compared with the NG group.
Increased Superoxide Anion Generation in Urinary Bladder Pericytes Exposed to High-Glucose Conditions
We performed hydroethidine staining to evaluate superoxide anion production in urinary bladder pericytes 48 hours after exposure to normal-glucose or high-glucose conditions. The generation of superoxide anion was considerably higher in urinary bladder pericytes exposed to high-glucose conditions than in those exposed to normal-glucose conditions (Fig. 5).
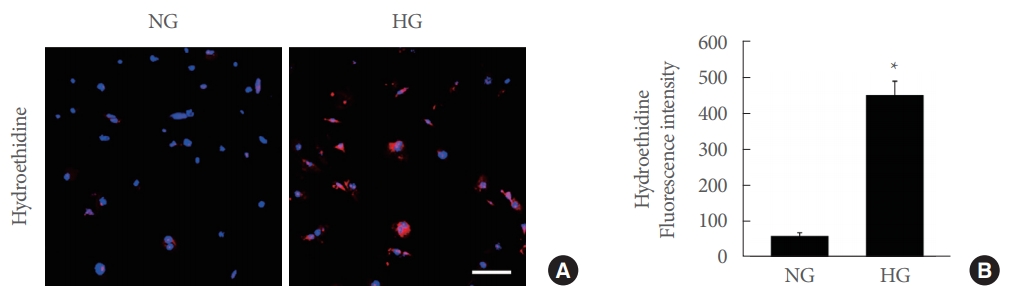
Increased superoxide anion generation in mouse urinary bladder pericytes exposed to high-glucose conditions. (A) In situ detection of superoxide anion. Pericytes were incubated under normal-glucose (NG; 5 mmol/L) or HG (30 mmol/L) conditions for 48 hours. Pericytes were incubated with hydroethidine, an oxidative fluorescent dye used to detect superoxide anion. Nuclei were labeled with the DNA dye 4,6-diamidino-2-phenylindole. Scale bar=100 μm. (B) The fluorescence intensity of hydroethidine was measured using NIH ImageJ software. Each bar represents the mean value (±standard error) of 6 separate wells per group. *P<0.05 compared with the NG group.
DISCUSSION
Here, we successfully isolated and cultured pericytes from the mouse urinary bladder. The morphologic features of the primary cultured urinary bladder pericytes were similar to those of cells isolated from human brain microvessels and mouse erectile tissues [17].
There is no single entirely pericyte-specific marker [15]. In the present study, therefore, we used PDGFR-β and NG2 as makers for pericytes, because these makers were useful to define pericytes from urinary bladder and erectile tissue [14,17,20].
Oxidative stress is one of the most important causes of diabetic complications in various organs [24,25]. In the present study, there was a significant increase in superoxide anion production when urinary bladder pericytes were exposed to highglucose conditions. A study has indicated that mitochondria are the main source of superoxide anion and peroxynitrite [26]. In agreement with our findings, high-glucose conditions have been reported to increase the generation of ROS in human urinary bladder smooth muscle cells in vitro [17]. We also observed that the exposure of urinary bladder pericytes to highglucose conditions promoted the apoptosis of the pericytes. Oxidative stress under high-glucose conditions has been found to activate the proapoptotic pathway in cultured detrusor muscle cells in vitro [27]. Previous studies have also reported that oxidative stress could induce the disruption of the detrusor muscle by enhancing smooth muscle cell apoptosis [28,29]. The generation of ROS in diabetes is known to inhibit neuronal survival and promote neurodegeneration in the urinary bladder [30]. Therefore, increased oxidative stress under high-glucose conditions may play a key role in the apoptosis of urinary bladder pericytes. However, we cannot exclude the possibility that high-glucose condition would also induce necroptosis of the pericytes.
In the present study, the urinary bladder pericytes themselves could contribute to well-formed tube-like structures under normal-glucose conditions, which was severely impaired when the cells were cultured under high-glucose conditions. In a previous study involving STZ-induced diabetic rats, it was found that the blood vessel and nerve fiber densities in the urinary bladder were decreased in a time-dependent manner [31]. We recently reported that pericytes cover the surface of endothelial cells in the suburothelial capillary plexus of the mouse urinary bladder. In contrast, large-diameter blood vessels are mainly covered with smooth muscle cells [14]. In agreement with these findings, the pre- and postcapillary microvessels and capillaries have been reported to be enclosed by pericytes [32].
Pericytes generate spontaneous Ca2+ transients and are involved in the phasic constriction of venules to maintain suburothelial microcirculation [33,34]. Disturbances in the constriction mechanisms of pericytes in capillary or postcapillary venules in diabetes may cause tissue hypoxia or blood flow stagnation [34]. Moreover, the diabetes-induced loss of pericytes may lead to extravasation from mucosal microvessels and result in neurogenic inflammation by stimulating chemosensitive afferent nerve fibers [35]. In addition, substance P-positive nerve fibers (primary afferent nerves) surrounding the microvasculature have been suggested to be involved in extravasation and neurogenic inflammation [36,37]. Previously, we demonstrated that the loss of pericyte function resulted in erectile function decline in normal mice, whereas the gain of pericyte function with hepatocyte growth factor restored erectile function in diabetic mice [17]. Further studies are needed to determine whether the loss or gain of pericyte function would affect urinary bladder functions.
From a technical point of view, great care must be taken to minimize contamination with epithelial cells. Although the urothelial layer was meticulously removed from the underlying submucosa and detrusor muscle layer by gentle swabbing with a Q-tip, it was not easy to completely avoid contamination with urothelial cells as shown in Fig. 1B. However, we could easily detect the morphologically distinct outgrowth of epithelial cells with the aid of a microscope, and this area was excluded from further subculture.
To the best of our knowledge, this is the first study to isolate and culture pericytes from the mouse urinary bladder. Currently, we are also conducting experiments to establish a protocol for isolating primary endothelial cells from the mouse urinary bladder. Although our in vitro model may not completely represent the complexity of in vivo DBD, we believe that these cellular models could clarify the role of pericyte-endothelial cell signaling in the physiology of normal micturition and the pathophysiology of DBD.
In summary, we established a protocol to isolate pericytes from the mouse urinary bladder and created an in vitro model of diabetic pericyte dysfunction to understand the role of this cellular component in the pathogenesis of DBD. This model would be a useful tool for screening the efficacy of therapeutic candidates targeting pericyte function in DBD and exploring the functional role of specific targets at the cellular level.
Notes
Fund/Grant Support
This work was supported by the National Research Foundation of Korea (NRF) grant (Ji-Kan Ryu [2019R1A2C2002414]) and by a Medical Research Center Grant (Ji-Kan Ryu, 2014R1A5A2009392) funded by the Korean government (Ministry of Science, ICT and Future Planning).
Research Ethics
The experimental protocol was approved by the Institutional Animal Care and Use Subcommittee of Inha University (approval number: INHA 190813-661).
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION STATEMENT
· Conceptualization: JS, JR
· Data curation: MC, NNM, KS, AL, KG, MK, JO, GNY
· Formal Analysis: MC, NNM, KS, AL, KG, MK, JO
· Funding acquisition: JS, JR
· Methodology: MC, NNM, K Song, KS, AL, KG, MK, JO, GNY
· Project Administration: JS, JR
· Visualization: MC, NNM, KS, AL, KG, MK, JO, GNY, JR
· Writing – Original Draft: MC, JS, JR
· Writing – Review & Editing: JS, JR