Methyl-CpG Binding Protein 2 in Alzheimer Dementia
Article information

Abstract
Despite decades of research on Alzheimer disease, understanding the complexity of the genetic and molecular interactions involved in its pathogenesis remains far from our grasp. Methyl-CpG Binding Protein 2 (MeCP2) is an important epigenetic regulator enriched in the brain, and recent findings have implicated MeCP2 as a crucial player in Alzheimer disease. Here, we provide comprehensive insights into the pathophysiological roles of MeCP2 in Alzheimer disease. In particular, we focus on how the alteration of MeCP2 expression can impact Alzheimer disease through risk genes, amyloid-β and tau pathology, cell death and neurodegeneration, and cellular senescence. We suggest that Alzheimer disease can be adversely affected by upregulated MeCP2-dependent repression of risk genes (MEF2C, ADAM10, and PM20D1), increased tau accumulation, and neurodegeneration through neuronal cell death (excitotoxicity and apoptosis). In addition, we propose that the progression of Alzheimer disease could be caused by reduced MeCP2-mediated enhancement of astrocytic and microglial senescence and consequent glial SASP (senescence-associated secretory phenotype)-dependent neuroinflammation. We surmise that any imbalance in MeCP2 function would accelerate or cause Alzheimer disease pathogenesis, implying that MeCP2 may be a potential drug target for the treatment and prevention of Alzheimer disease.
ALZHEIMER DISEASE
Alzheimer disease is a neurodegenerative disease that constitutes the most common form of dementia without a cure. Alzheimer’s Association Reports projected that the social and economic burden and the global prevalence of Alzheimer disease will grow constantly until 2050 [1], demanding immediate attention to research on the treatment and prevention of Alzheimer disease. The pathogenesis of Alzheimer disease is defined by the neurotoxic accumulation of extracellular amyloid-β plaques and intracellular tau neurofibrillary tangles, glial scarring and gliosis, and aberrant alterations in synaptic plasticity, ultimately resulting in severe cognitive impairments and extensive neuronal loss [2]. The cognitive dysfunctions observed in Alzheimer disease primarily consist of memory loss and problems in language or thinking skills.
Among the diverse causes of Alzheimer disease identified to date, the foremost studies have revealed that Alzheimer disease can arise from familial or sporadic mutations in a number of risk genes, most notably amyloid precursor protein (APP), presenilin-1 or -2 (PSEN1, PSEN2), β-secretase 1 (BACE1), and apolipoprotein E (APOE) [3]. Research on these risk genes mainly revolved around the amyloid hypothesis, positing that dysfunction in the processing of amyloid-β protein—more specifically, the imbalance between production and clearance of amyloid-β peptides such as amyloid-β-42—is the essential cause of Alzheimer disease [2]. However, recent failures of amyloid-β monoclonal antibodies in clinical trials warrant the identification of new genetic and molecular targets for the treatment and prevention of Alzheimer disease [4,5]. For instance, recent efforts from largecohort genome-wide association studies (GWAS) have discovered a number of novel risk genes for Alzheimer disease [6,7] and researchers are bringing tau pathology into the spotlight in Alzheimer disease research [8,9].
Notably, mounting evidence and recent breakthroughs have highlighted neuroinflammation as a critical part of the pathogenesis of Alzheimer disease [10,11]. Pathological neuroinflammation in Alzheimer disease is principally initiated by amyloid-β-dependent, tau-dependent, or idiopathic long-lasting activation of microglia, the proficient innate immune cells in the central nervous system. Subsequently, the sustained release of proinflammatory and neurotoxic mediators leads to an increased emergence of reactive astrocytes and abnormal accumulation of oxidative stress in the brain, ultimately rendering the brain microenvironment inhospitable to neurons. Neuroinflammation is therefore thought to be a central factor underlying the process of neurodegeneration in Alzheimer disease [12].
It is also important to note that a potential contributor to neuroinflammation in the aging brain is cellular senescence, which is apt to develop in replication-competent glial cells [13]. Cellular senescence is characterized by irreversible cell cycle arrest, resistance to apoptosis, and the senescence-associated secretory phenotype (SASP). Interestingly, the SASP converts senescent brain cells into a pro-inflammatory phenotype that releases cytokines, chemokines, proteases, and growth factors [14], which in turn can activate neighboring microglia and astrocytes to give rise to reactive glia. Through this mechanism, cellular senescence can prompt neuroinflammation in the brain through both reactive glia and senescent glia. In addition, recent evidence has demonstrated that the accumulation of senescent glial cells is critical for the manifestation of Alzheimer disease phenotypes, including neurodegeneration and cognitive decline [15,16]. These findings jointly suggest that cellular senescence may be the cellular intermediary connecting molecular pathology to the behavioral phenotype of Alzheimer disease.
METHYL-CpG BINDING PROTEIN 2
Methyl-CpG binding protein 2 (MeCP2) is a chromosomal protein that recognizes and binds to methylated CpG nucleotide sequences to control transcription via epigenetic modulation. The main modes of transcriptional regulation by MeCP2 include histone deacetylase-dependent histone deacetylation, bridging histone methylation and DNA methylation, and CREB1-dependent transcriptional activation [17-20]. MeCP2 is known to be critical for the normal functioning of the nervous system, as it can selectively repress the expression of the BDNF gene [21,22] and shape multiple aspects of excitatory and inhibitory neurotransmission in diverse manners [23-25]. In addition, deficient MeCP2 functioning leads to impaired neuronal maturation in the brain [26,27] and the X-linked progressive neurodevelopmental disorder Rett syndrome, which is characterized by severe mental retardation and autistic features, mainly in females [28,29].
Interestingly, a study revealed that hippocampal MeCP2 was upregulated in Alzheimer disease patients [30], but another study showed that MeCP2 expression was reduced in the hippocampus of APP/PS1 mice and that upregulation of hippocampal MeCP2 could play a therapeutic role in an Alzheimer disease mouse model [31]. These data demonstrate that there exists an interaction between Alzheimer disease and MeCP2, which requires an in-depth investigation. This mini-review aims to identify the common functional traits between MeCP2 and biological factors of Alzheimer disease, and to summarize findings from recent studies in order to propose hypotheses regarding the pathophysiological roles of MeCP2 in Alzheimer disease. Specifically, we focus on the interactions of MeCP2 with Alzheimer disease risk genes and amyloid-β and tau pathology, as well as the MeCP2-dependent regulation of neuronal cell death and cellular senescence.
MeCP2 AND ALZHEIMER DISEASE RISK GENES
In the past decades, genetic association studies have revealed a multitude of risk genes for Alzheimer disease, but only a handful can be linked to MeCP2. The MeCP2-linked Alzheimer disease risk genes include MEF2C, ADAM10, and PM20D1.
MeCP2 Represses MEF2C
MEF2C encodes a transcriptional factor (myocyte enhancer factor 2C; Mef2c) that is involved in cardiac, vascular, and neuronal development [32-34]. In the brain, MEF2C plays a critical role by regulating neurogenesis, neuronal survival, and synaptic plasticity [35,36]. Mutations in MEF2C lead to neurodevelopmental disabilities including severe mental retardation [37]. In the largest GWAS of late-onset Alzheimer disease (LOAD) to date [6,7], MEF2C has been identified as a potential risk gene for Alzheimer disease, but the pathophysiological role of MEF2C in Alzheimer disease remains unclear.
MeCP2 binds to the promotor region of the MEF2C gene and represses Mef2c expression in the mouse brain and humanderived cells [20,38]. Since MeCP2 expression is elevated in the brain of individuals with Alzheimer disease and MEF2C dysfunction leads to mental retardation, MeCP2-mediated repression of Mef2c could contribute to cognitive decline in Alzheimer disease.
MeCP2 Represses ADAM10
ADAM10, which encodes ADAM metallopeptidase domain 10, is responsible for the proteolytic cleavage of a number of cellsurface proteins. Notably, the product of ADAM10 functions as an α-secretase involved in cleaving the ectodomain of amyloid precursor protein (APP) [39]. Rare variants of ADAM10 have been found to cosegregate in families affected by LOAD [40,41]. In addition, the largest GWAS of LOAD have identified ADAM10 as a risk gene for Alzheimer disease [6,7]. ADAM10 variants can increase amyloid-β levels in vitro [40], and have been found to induce amyloidogenic cleavage and increase the amyloid-β plaque load in an Alzheimer disease mouse model [41]. However, APP cleavage by wild-type α-secretase is thought to be neuroprotective in Alzheimer disease since wild-type α-secretase cleaves a site within the amyloid-β sequence to prevent the overall production of amyloid-β [42].
A recent study demonstrated that MeCP2 inhibited ADAM10 expression in mouse neural progenitor cells and human-derived cells [43]. Since Alzheimer disease patients could carry rare variants of ADAM10, MeCP2-dependent inhibition of ADAM10 could disrupt the activity of mutant α-secretase, thereby reducing amyloid-β deposition in the brain in Alzheimer disease patients. However, MeCP2-dependent inhibition of ADAM10 could also increase the amyloid-β load in Alzheimer disease patients who retain wild-type ADAM10.
MeCP2 Represses PM20D1
The product of peptidase M20 domain containing 1 (PM20D1) is a circulating biosynthetic enzyme that catalyzes the condensation of fatty acids and a number of amino acids to produce Nacyl amino acids [44]. Increased levels of PM20D1 promote energy expenditure in vivo. However, the role of PM20D1 in neurons and in the brain remains unclear. A methylated quantitative trait locus study revealed that PM20D1 was the sole risk gene with consistently enriched promoter hypermethylation in Alzheimer disease patients [45]. Moreover, PM20D1 levels increased after neurotoxic insults in an APP/PS1 mouse model and human Alzheimer disease patients, and PM20D1 was shown to protect neurons against the insults. Increasing or decreasing PM20D1 expression reduces or aggravates Alzheimer disease pathology, respectively. These data suggest that PM20D1 plays a neuroprotective role against Alzheimer disease.
Interestingly, increased binding of MeCP2 to the PM20D1 promoter was observed in immortalized B cells from Alzheimer disease patients with the methylated rs708727-rs960603 haplotype, and the MeCP2-bound promoter of PM20D1 showed decreased histone acetylation [45]. These data suggest that MeCP2 inhibits PM20D1 via induction of a transcriptionally silent chromatin state. Thus, MeCP2-PM20D1 interactions could render the brain susceptible to neurodegeneration and cognitive decline in response to neurotoxic mediators of Alzheimer disease pathology.
MeCP2 Is Unlikely to Be Linked With PSEN and BACE1
PSEN1 and PSEN2 encode presenilin-1 and presenilin-2, which form the core catalytic subunit of γ-secretase, whereas BACE1 encodes β-secretase [42]. The γ- and β-secretases sequentially cleave APP to produce amyloid-β. Mutations in PSEN genes can cause incomplete digestion of amyloid-β, which leads to enhanced amyloid-β-42 production, and could also lead to a loss of other essential presenilin functions and trigger the cellular and behavioral impairments found in Alzheimer disease.
The links between MeCP2 and the PSEN genes and BACE1 are weak. MeCP2 expression was not correlated with the PSEN1 or PSEN2 expression profile in the developing cerebral cortex of wild-type mice [46]. Mice with MeCP2 ablation under transverse aortic constriction exhibited enrichment of the PSEN pathway in a bioinformatics analysis [47], but the lack of more direct evidence precludes further interpretation. In addition, BACE1 knockdown did not affect MeCP2 expression in human-derived cells [48]. These data indicate that the PSEN genes and BACE1 are unlikely to control MeCP2 expression.
MeCP2 and Alzheimer Disease Risk Genes: Summary
MeCP2 can repress the novel Alzheimer disease risk genes MEF2C, ADAM10, and PM20D1. Each interaction could differentially impact Alzheimer disease. MEF2C repression by MeCP2 could induce deficits in neuronal functions that lead to cognitive decline. ADAM10 inhibition by MeCP2 could bidirectionally impact the amyloid-β load in the brain depending on the presence of variations in the ADAM10 gene. PM20D1 repression by MeCP2 could impair the intrinsic neuroprotection mechanism against Alzheimer disease. However, the traditionally acknowledged risk genes of Alzheimer disease—the PSEN genes and BACE1—may not be necessarily correlated with MeCP2.
Overall, these views indicate that increased MeCP2 in the brain could influence the pathology and phenotype of Alzheimer disease in diverse manners, most likely through interactions with recently discovered Alzheimer disease risk genes. However, research on MeCP2 and Alzheimer disease should account for the potentially unidentified interactions between MeCP2 and other risk genes of Alzheimer disease, as well as genes indirectly related to Alzheimer disease. Moreover, it should be noted that age-specific and cell type-specific interactions of MeCP2 with Alzheimer disease risk genes may exert a differential influence on the pathophysiology of Alzheimer disease. Finally, the directionality of interactions between MeCP2 and Alzheimer disease risk genes should be considered, such that mutations in the PSEN genes and BACE1 may not affect MeCP2, but MeCP2 could control the expression of the PSEN genes, BACE1, and other classical risk genes of Alzheimer disease.
MeCP2 AND THE PATHOLOGY OF ALZHEIMER DISEASE
Amyloid-β and tau are the building blocks of amyloid plaque and neurofibrillary tangle; the most widely recognized pathological manifestations of Alzheimer disease. Accumulation levels of amyloid-β and tau are frequently used as a scale to measure the progress of Alzheimer disease. Here, we review recent studies that explored the correlation between aberrant expression of MeCP2 and the pathology of Alzheimer disease.
The Relationship Between MeCP2 and Amyloid Pathology Is Controversial
MeCP2 expression and phosphorylation levels were increased in the hippocampal CA1 of amyloid-β-injected rats [49]. In addition, MeCP2 siRNA treatment restored hippocampal dendritic spine numbers and memory deficits in a Morris water maze in amyloid-β-injected rats. However, amyloid-β was shown to reduce MeCP2 expression in immortalized hippocampal neurons and in the hippocampus of APP/PS1 mice [31], in contrast with the above finding. In addition, MeCP2 deficiency did not alter the level of APP in the cortex, although it disturbed the localization of APP to the cell membrane [50].
MeCP2 Controls Tau Pathology and Vice Versa
In a transgenic mouse model of tauopathy overexpressing the human tau gene, whole genome expression analysis revealed that both total and phosphorylated MeCP2 were upregulated in the hippocampus [51]. Total MeCP2 levels were also upregulated in the cortex and cultured cortical neurons from the tauopathy mouse model. Importantly, human Alzheimer disease brain tissue also exhibited elevated levels of total and phosphorylated MeCP2, which is consistent with the findings from the tauopathy mice model. Furthermore, mice with MeCP2 triplication syndrome displayed elevated tau expression within the hippocampus and cortex, followed by neuronal loss in these brain regions [52]. Cognitive impairment was also evident in Y-maze and novel object recognition tests.
In vitro, MeCP2 knockdown in a human tau-expressing neuronal cell line also resulted in significantly decreased levels of total and phosphorylated tau [51].
MeCP2 and the Pathology of Alzheimer Disease: Summary
The equivocal findings in the literature suggest that the link between MeCP2 and amyloid-β remains unclear. Nonetheless, increased levels of tau consistently resulted in both the overexpression and activation of MeCP2, and alterations in MeCP2 expression were positively correlated with tau levels in neurons. From these data, a critical connection between MeCP2 and Alzheimer disease tau pathology is apparent. In the future, more in-depth studies are needed to reveal the causal relationship and modes of interaction between MeCP2 and tau pathology.
MeCP2 AND NEURONAL CELL DEATH: APOPTOSIS, EXCITOTOXICITY, AND NEURODEGENERATION
MeCP2 is involved in the regulation of neuronal cell death by apoptosis and excitotoxicity. Although the precise mechanism underlying neurodegeneration in Alzheimer disease is unclear, studies have demonstrated that in Alzheimer disease, the brain is afflicted by increased excitotoxic and apoptotic cell death [53,54], suggesting that both excitotoxicity and apoptosis can contribute to the neurodegenerative process in Alzheimer disease. In this instance, alteration in MeCP2 expression may directly mediate neurodegeneration through excitotoxicity and apoptosis.
Increased MeCP2 Dosage Leads to Excitotoxicity and Apoptosis
In cultured neurons and neuronal tissue, MeCP2 overexpression increases apoptotic cell death [55,56] (but also see [57]). MeCP2 overexpression-dependent apoptosis could arise from excitotoxicity mediated by astrocytic glutamate release [52]. More importantly, an increase in the MeCP2-e2 isoform was found to mediate amyloid-β-induced apoptosis in cultured cortical neurons [58], demonstrating the potential significance of MeCP2 for neurodegeneration in Alzheimer disease.
In vivo, a human MeCP2-e2 isoform gain-of-function mutation promoted apoptosis in Drosophila motor neurons [59]. In addition, MeCP2 overexpression led to neuronal loss in the hippocampus, cortex, and cerebellum in a MeCP2 triplication syndrome mouse model [52].
Decreased MeCP2 Dosage Facilitates Cell Death
In cultured neurons, MeCP2 deficiency resulted in a greater magnitude and faster onset of apoptotic cell death when the neurons were exposed to either excitotoxic or hypoxic-ischemic insults [60]. However, although microglial MeCP2 knockdown directly caused NMDA receptor-dependent excitotoxic neuronal cell death [61], astrocytic MeCP2 deficiency did not affect the rate of neuronal cell death [62].
In vivo, MeCP2 deficiency resulted in a reduced number of medullar tyrosine hydroxylase-expressing neurons, but the effect was not due to apoptosis [63], and MeCP2 deficiency had no effect on apoptotic cell death in the locus coeruleus [64,65]. However, MeCP2 deficiency increased apoptosis in immature olfactory neurons in mice at 52 weeks of age, but not at 12 weeks of age or younger [66]. In addition, degenerating dopaminergic neurons in mice exhibited reduced MeCP2 activity [67].
MeCP2 and Neuronal Cell Death: Summary
Increased MeCP2 expression can directly induce excitotoxicity and apoptosis, which could underlie amyloid-β-induced apoptotic neurodegeneration. In the future, research on the roles of the MeCP2-e2 isoform and astrocytic MeCP2 could lead to a broader understanding of the biological pathways underlying neurodegeneration in Alzheimer disease.
However, MeCP2 deficiency does not seem to directly impact neuronal cell death. Overall MeCP2 downregulation may just indirectly facilitate neuronal cell death by rendering neurons susceptible to cell death-triggering insults or events (e.g., excitotoxicity, hypoxia-ischemia, aging, or neurodegenerative disease), although microglial MeCP2 reduction seems to directly dysregulate neuronal cell survival through excitotoxicity. Further studies are required to clarify the link between MeCP2 deficiency and neuronal cell death.
MeCP2 AND CELLULAR SENESCENCE
Cellular senescence is thought to be one of the underlying causes of aging [68], promoting aging-related phenotypes [69,70] and contributing to the pathogenesis of neurodegenerative diseases including Alzheimer disease [15,16]. Interestingly, accumulating evidence indicates that MeCP2 is associated with cellular senescence, which suggests cellular senescence as a potential linker between MeCP2 and Alzheimer disease.
The Link Between Increased MeCP2 Levels and Cellular Senescence Is Elusive
Human embryonic lung fibroblasts undergoing senescence exhibited upregulation of MeCP2 mRNA along with increased senescence-associated β-galactosidase (SA-β-gal) activity, but chemically induced premature senescence did not change the expression of MeCP2 in these cells [71]. In addition, MeCP2 protein levels did not differ between lung fibroblasts with lower and higher degrees of senescence [72]. Moreover, MeCP2 overexpression-induced senescence in endothelial progenitor cells resulted in an increased number of apoptosis cells and cleaved caspase-3 protein levels [73], in contrast to the apoptosis-resistant characteristic of senescent cells.
MeCP2 Downregulation Induces Cellular Senescence
Neural progenitor cells and neurons differentiated from fibroblasts of patients with Rett syndrome displayed elevated SA-β-gal activity, p53 induction, and altered SASP signature genes [74]. Furthermore, mesenchymal stem cells from Rett syndrome patients showed a lower degree of apoptosis and were prone to senescence, which was accompanied by a reduction of apoptosis-related genes and upregulation of senescence-promoting genes [75-77]. Furthermore, partial silencing of MeCP2 in human mesenchymal stem cells reduced apoptosis and triggered senescence [78].
MeCP2 and Cellular Senescence: Summary
The relationship between MeCP2 overexpression and senescence seems controversial since the results are generally equivocal. In contrast, MeCP2 downregulation was significantly associated with cellular senescence. MeCP2-silenced cells and Rett syndrome patient-derived cells consistently exhibited a senescent phenotype, alterations in senescence-related genes, and decreased apoptosis. Since cellular senescence affects the major pathology of neurodegenerative diseases, the correlation between Alzheimer disease and cellular senescence induced by MeCP2 downregulation may be worthy of further investigation.
MeCP2 and Urological Dysfunction
Earlier reviews of Rett Syndrome considered the aspect of its recurrent infection a quandary [79]. Mixed with symptoms of meningitis, urinary tract infections have been considered a peripheral occurrence, not deeply associated with the pathognomonic representation.
Link between Rett syndrome and urinary dysfunction
Earlier studies of MeCP2 and its association with the urinary tract has been limited to familial studies. As in Rett syndrome, considered to be exclusively female, the association with the MECP2 gene has identified male cases which involve Klinefelter’s mosaic, males with severe neonatal encephalopathy in families with Rett syndrome and males who are mosaic for the MECP2 mutation [80]. Zeev et al. [80] describes male mutation in familial Rett syndrome where it manifests as bladder and ureteral hypotonicity, resulting in early and recurrent urinary tract infections. In contrast to female involvement of predominant constipation, breathing abnormalities and cardiac arrhythmias as the manifestation of its autonomic nervous system involvement.
MeCP2 involving peripheral neural and smooth muscle plasticity
Recent advances have allowed review of the genetic data of Rett syndrome patients, positively identifying the association between MeCP2 and urological dysfunction [81]. Other studies have shown miR-132 and miR212 involving synaptic plasticity and cholinergic signaling affected outside the CNS, including smooth muscle, and changes in MeCP2 [82]. In this study, bladder outlet obstruction induced miR-132/212 and repressed MeCP2 and cell viability. Other studies have also shown miR-132 overexpression in bladder overactivity as well [83].
HYPOTHESES AND CONCLUSION
Studies have demonstrated elevated MeCP2 levels in the cortex and hippocampus of Alzheimer disease patients. Aberrantly increased MeCP2 could exacerbate Alzheimer disease through multiple mechanisms. First, the MeCP2-mediated repression of the Alzheimer disease risk genes MEF2C, ADAM10, and PM20D1 could negatively impact the progression of Alzheimer disease. Second, MeCP2 could drive the accumulation of tau protein in the brain. Lastly, MeCP2 could render neurons prone to neurodegeneration by inducing excitotoxicity and apoptosis. In the future, corroborating evidence should be sought and the underlying mechanisms should be determined to clarify the roles of increased MeCP2 in Alzheimer disease.
However, although MeCP2 levels were found to be elevated in the brain of Alzheimer disease patients, its cell type-specific expression pattern in Alzheimer disease has not been documented to date. Studies have demonstrated that MeCP2 deficiency can induce cellular senescence, while senolytic therapy—selective elimination of senescent cells—is effective against astrocyte- and microglia-induced defects in Alzheimer disease [15,16]. Synthesizing these observations, in Alzheimer disease, the brain may exhibit an abnormally large extent of astrocytic and microglial senescence, potentially due to MeCP2 downregulation in the glia, which in turn can accelerate neuronal dysfunction and neurodegeneration in Alzheimer disease through glial SASP. Future studies should elucidate the cell type-specific expression and functions of MeCP2 in brain tissue affected by Alzheimer disease, along with the pathophysiological roles of reduced MeCP2 in Alzheimer disease-associated senescence.
Integrating the data handled in this review, we suggest a model in which Alzheimer disease pathogenesis could be exacerbated by MeCP2 dysfunction (Fig. 1). MeCP2 upregulation could dysregulate Alzheimer risk genes and accelerate tau pathology, while MeCP2 deficiency could induce glial senescence that contributes to neuroinflammation and excitotoxicity in Alzheimer disease. In both pathways, MeCP2 dysfunction-induced neuronal cell death and neurodegeneration play a major role.
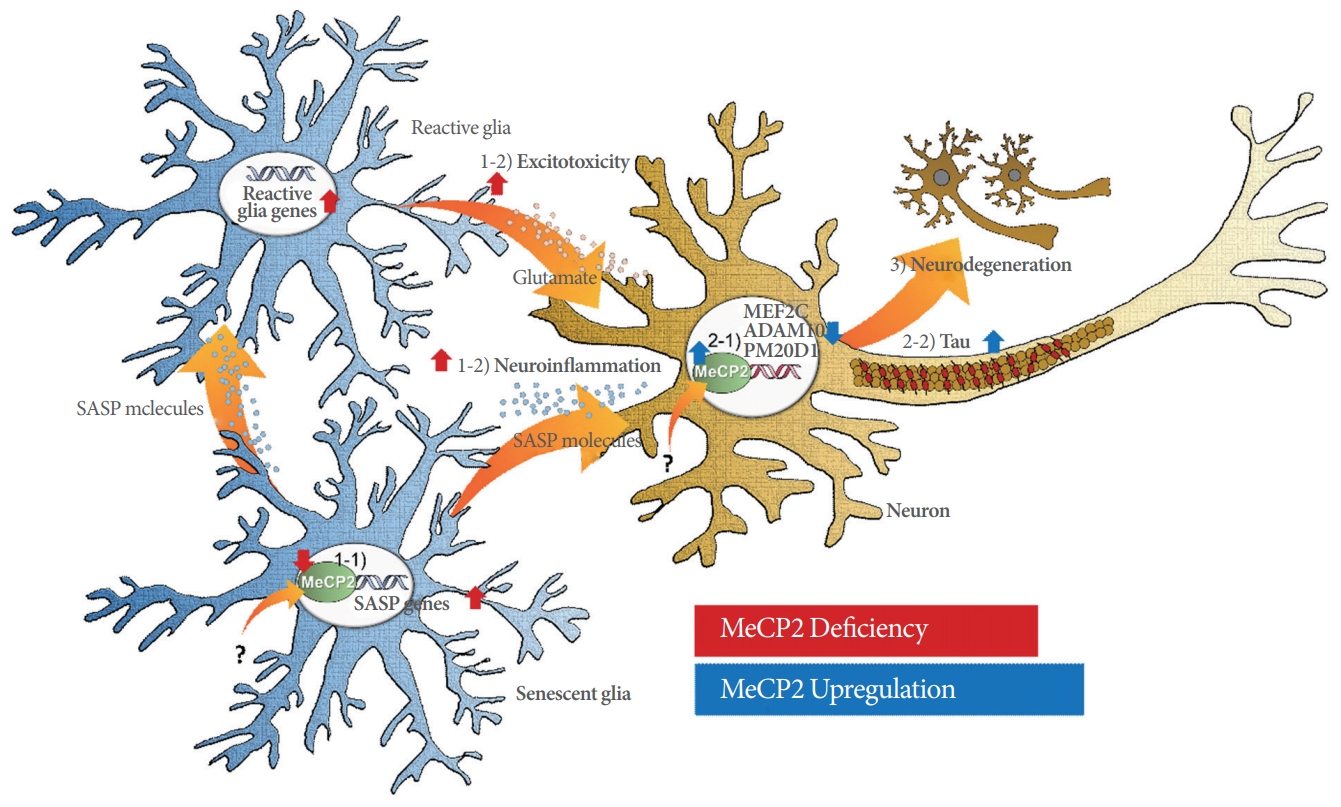
A schematic model depicting the impact of MeCP2 dysfunction in Alzheimer disease. 1-1) MeCP2 deficiency induces cellular senescence and upregulation of senescence-associated secretory phenotype (SASP) genes in glial cells (astrocytes and microglia). 1-2) Senescent glia secrete SASP molecules (cytokines, chemokines, proteases, and growth factors), which in turn directly induce neuroinflammation in neurons and give rise to reactive glia that induce excitotoxicity. 2-1) MeCP2 upregulation causes downregulation of the Alzheimer disease risk genes MEF2C, ADAM10, and PM20D1 in neurons. 2-2) In conjunction, MeCP2 upregulation exacerbates tau pathology in neurons. 3) Taken together, MeCP2 dysfunction results in the neurodegeneration observed in Alzheimer disease. Red arrows indicate the effects exerted by MeCP2 deficiency and blue arrows indicate the effects resulting from MeCP2 upregulation.
Research from the recent decade has suggested that MeCP2 could play a multifaceted role in Alzheimer disease. From the mounting evidence, we propose that MeCP2 can repress Alzheimer disease risk genes, prevent the aggravation of tau pathology, and inhibit the induction of neuronal cell death and cellular senescence. Further studies of the genetic and molecular mechanisms underlying the impact of MeCP2 on Alzheimer disease should follow in order to reveal the therapeutic potential of MeCP2 as a drug target for Alzheimer disease and provide novel insights into the pathogenesis of Alzheimer disease.
Notes
Fund/Grant Support
This work was supported by the Korea Ministry of Science & ICT (18-BR-03-02) and the National Research Council of Science & Technology (NST) by the Korean government (MSIP) (No. CRC-15-04-KIST).
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION STATEMENT
·Full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis: BK, HII
·Study concept and design: BK, YC, HII
·Acquisition of data: BK, YC
·Analysis and interpretation of data: BK, YC
·Drafting of the manuscript: BK, YC
·Critical revision of the manuscript for important intellectual content: BK, YC
·Statistical analysis: BK, YC
·Obtained funding: HII
·Administrative, technical, or material support: HSK
·Study supervision: HII