Changes in Synaptic Transmission and Long-term Potentiation Induction as a Possible Mechanism for Learning Disability in an Animal Model of Multiple Sclerosis
Article information

Abstract
Purpose:
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the central nervous system. It has been shown that memory deficits is common in patients with MS. Recent studies using experimental autoimmune encephalomyelitis (EAE) as an animal model of MS have shown that indicated that EAE causes hippocampal-dependent impairment in learning and memory. Thus far, there have been no in vivo electrophysiological reports describing synaptic transmission in EAE animals. The aim of the present work is to evaluate the synaptic changes in the CA1 region of the hippocampus of EAE rats.
Methods:
To evaluate changes in synaptic transmission in the CA1 region of the hippocampus of EAE rats, field excitatory postsynaptic potentials (fEPSPs) from the stratum radiatum of CA1 neurons, were recorded following Schaffer collateral stimulation.
Results:
The results showed that EAE causes deficits in synaptic transmission and long-term potentiation (LTP) in the hippocampus. In addition, paired-pulse index with a 120 msec interstimulus interval was decreased in the EAE group. These findings indicate that EAE might induce suppression in synaptic transmission and LTP by increasing the inhibitory effect of GABAB receptors on the glutamate-mediated EPSP.
Conclusions:
In conclusion, influence of inflammation-triggered mechanisms on synaptic transmission may explain the negative effect of EAE on learning abilities in rats.
INTRODUCTION
Multiple sclerosis (MS) is a chronic inflammatory disease of the nervous system [1]. During this disease, lymphocytic infiltration into the central nervous system (CNS) results in the demyelination and damage of axons. Between 40% and 60% of MS patients experience cognitive deficits such as learning and memory dysfunction [2]. Loss of cognitive function is one of the earliest manifestations of MS [3]. The mechanisms underlying these cognitive deficits remain poorly understood. It has been demonstrated that CNS inflammation could influence the induction of neuroplasticity and the function of neuronal networks, such as synaptic plasticity [4]. Postmortem hippocampal sections from MS patients show demyelination and neuropathology in the hippocampus [5]. Furthermore, hippocampal CA1 atrophy was reported in MS patients [6] and in an MS animal model [7].
Experimental autoimmune encephalomyelitis (EAE) is one of the best-studied animal models for MS. It is characterized by motor symptoms and cognitive impairments. Recently, Di Filippo et al. [8] reported that EAE could block the induction of long-term potentiation (LTP) in hippocampal slice preparations from mice. However, it is unclear whether EAE could effect in vivo LTP induction.
Thus, the aim of this study was to evaluate in vivo the effect of EAE on synaptic transmission, and LTP induced by the high frequency stimulation (HFS) of Schaffer collaterals in the CA1 region of the rat hippocampus. In addition, we used pairedpulse stimulation to determine whether the induction of EAE could alter inhibition or neurotransmitter release in the CA1 region of the hippocampal circuits.
MATERIALS AND METHODS
Animals
Forty male Wistar rats weighing 150 to 200 g were divided into 4 groups (2 control groups and 2 EAE groups) to measure synaptic transmission and LTP induction. The animals were housed under environmentally controlled conditions (12-hour light/dark cycles, temperature between 22°C±2°C) at the Arak University of Medical Sciences animal facility. Food and water were supplied ad libitum. All procedures were carried out in accordance with EU Directive 2010/63/EU and all regulations established by the Arak University of Medical Sciences Research Ethics Committee (# 92-159-11).
Induction of EAE
EAE was induced by the subcutaneous injection of 100-μg myelin basic protein (MBP) from guinea pig brain (Sigma Aldrich, St. Louis, MO, USA). The MBP was dissolved in 150 μL of phosphate buffered saline and emulsified in an equal volume of complete Freund’s adjuvant (Difco, BD, Franklin Lakes, NJ, USA) that included 0.2 mg of Mycobacterium tuberculosis H37Ra (Difco). The anesthetized animals were injected on their hind limbs with MBP on day 0 and day 7. Two intraperitoneal injections (IP) of 200-ng pertussis toxin (Sigma Aldrich) were administered on day 0 and day 2 (postimmunization). Control animals were injected with vehicle solution. All animals were evaluated daily for clinical signs using the following clinical scale: 0 (no clinical signs), 1 (partial loss of tail tonus), 2 (tail paralysis), 3 (paraparesis of hindlimb), 4 (paraplegia), 5 (tetraparesis), 6 (tetraplegia), and 7 (death).
Electrophysiology
Electrode positions
Electrophysiology recording was initiated 28 hours after an animal obtained a score of at least 3. Animals were anesthetized with urethane (1.2 g/kg, IP) and placed in the stereotaxic frame. Throughout the experiment, the rectal temperature was monitored and maintained at 36°C–37°C with a warming blanket. To stimulate the Schaffer collateral and commissural fibers, we used a twisted bipolar Teflon-coated stainless steel stimulating electrode (200-μm shaft diameter, A-M system, Carlsborg, WA, USA). We lowered the electrode vertically into the brain at 3.8 mm posterior and 2.8 mm lateral to bregma, through the dorsal hippocampus (depth, 2.8–3.4 mm). To record field potentials from the CA1 region of the hippocampus, we used a single Teflon-coated stainless steel electrode. The electrode was lowered vertically at 3.8 mm posterior and 2.0 mm lateral to bregma, through the dorsal hippocampus (depth, 2.8–3.4 mm). At the end of each experiment, the position of the electrodes was confirmed histologically.
Stimulation and recording
Electrophysiological (field potential) responses were recorded at the stratum pyramidale following stimulation of Schaffer collaterals. Electrical stimulation was supplied by a stimulus isolator unit (World Precision Instruments, Sarasota, FL, USA) and consisted of brief (200 μsec) constant current square pulses. The average evoked field potentials were required to be stable for 60 minutes before tetanus delivery. When the variation in field population spike (PS) amplitude was less than ±10% for 60 minutes, the baseline recording was considered stable. Animals that did not have stable baseline responses were excluded from the study. The response threshold was defined as the lowest stimulus intensity producing a consistently identifiable response. The stimulus intensity was then set to evoke a synaptic potential of a half-maximal PS. This stimulus intensity was also used for induction of LTP and to measure paired-pulse index (PPI). The PS amplitude was calculated as the average of the two potential differences of the negative spike peak to the preceding and following positive peaks.
For LTP induction, the tetanus consisted of 5 trains of 100 Hz; each train lasting 200 msec with an intertrain interval of 10 seconds. After tetanus, the response was recorded for 60 minutes.
The PPI was established from the mean percent change in PS. The PPI was calculated as follows: (P2/P1) where P2 is the amplitude of the second PS and P1 is the amplitude of the first PS. The paired-pulse protocol averaged 5 responses with an interval of 60 seconds. The synaptic connections of the hippocampus produce two distinct phases of the PPI depending on the interstimulus interval (ISI), an early inhibitory phase (20–50 msec), and a late facilitation phase (50–150 msec) [9]. In our study, we chose 20 msec ISI as the early phase and 120 msec ISI as the late phase.
Data Analysis
We averaged 5 responses at each stimulus intensity level. The averaged waveform was used for further analysis. The PS amplitude (mV) was determined from the averaged field potentials recorded from the stratum pyramidale. These signals were amplified (World Precision Instruments) and saved at a 10-kHz sampling rate on a personal computer hard disk for off-line analysis (WSI, Tehran, Iran). We acquired the PS amplitude change over time mediated by HFS with respect to the 10 minutes baseline recording. The data was presented as mean±standard deviation. Unpaired or paired t-tests were used when two independent or dependent groups were compared, respectively. Differences were considered significant at P<0.05.
RESULTS
Synaptic Transmission
Applying single pulses at 0.1 Hz to the Schaffer collateral inputs of CA1 neurons evoked a single PS in control and EAE rats. The CA1 field responses were increased in amplitude in proportion to the increase in stimulus intensity. The baseline responses for the control and EAE groups were stable at each stimulus intensity level with a stimulation rate of 0.1 Hz. The mean values of the baseline PS amplitude for control and EAE groups are shown in Fig. 1. As shown in Fig. 1, the mean PS amplitude in EAE animals (1.35 mV) are significantly lower than in the control rats (2.46 mV) (Unpaired t-tests, P=0.042, n=11).

Experimental autoimmune encephalomyelitis (EAE)-mediated inhibition of CA1-synaptic transmission. The population spike amplitude in control (n=11) and EAE (n=11) at low stimulus intensity. *P=0.042, Unpaired t-test.
Long-term Potentiation
To understand the effect of EAE on synaptic plasticity, we analyzed LTP in the CA1 region. After recording stable field excitatory postsynaptic potentials (fEPSPs) from the CA1 region, a tetanic stimulation was applied to the Schaffer collaterals. Fig. 2 shows the time course change in PS amplitude. The amplitude of the PS in control rats increased to 142.5%±15% of the baseline (n=11) when measured 60 minutes after tetanic stimulation, which is indicative of LTP induction. Induction of EAE completely blocked the PS potentiation (95.5%±10.5%, n=11), thus indicating that EAE could block LTP induction in the hippocampal CA1 region. Representative averaged field potentials at the pyramidal cell layer in control and EAE rats are shown in Fig. 3.
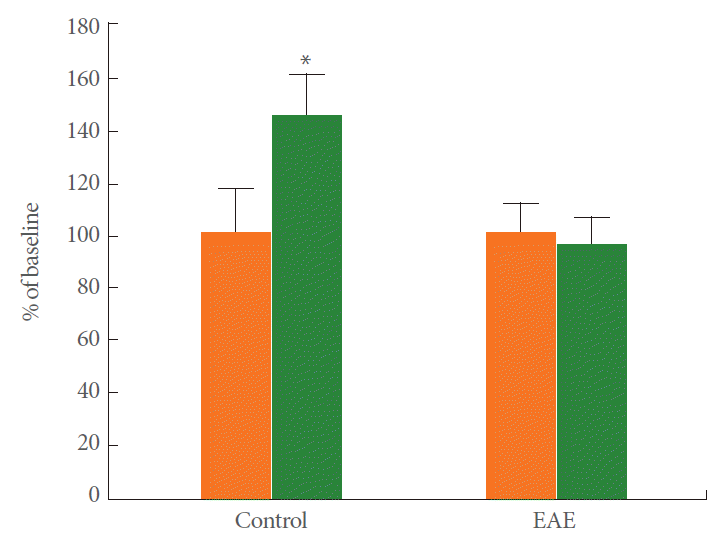
Induction of population spike (PS) long-term potentiation (LTP) in control and experimental autoimmune encephalomyelitis (EAE) rats. The PS amplitude change vs. time in control (n=11) and EAE (n=11) rats at the test stimulus intensity. Notice high frequency stimulation (HFS) induced PS LTP reversal 60 minutes after HFS in control but not in EAE rats. *P=0.001, Paired t-test.

Long-term potentiation is impaired in the Schaffer Collateral-CA1 synapses of the hippocampus in experimental autoimmune encephalomyelitis (EAE) rats, field population spike amplitude was plotted as percent of pretetanus baseline. Each point represents the mean±standard error of the mean. Right panel, representative averaged field potentials at the pyramidal cell layer in the control and EAE rats.
Paired-Pulse Index
To assess the impact of EAE on CA1 excitability at the circuit level, we determined the PPI of both animal groups using paired-pulse stimulation. Significant inhibition of the second population response was apparent at a 20 msec interpulse interval in control (n=10) and in EAE (n=8) rats. A comparison of the PPI in the 2 groups showed that with a 20 msec interpulse interval, the PPI was decreased in EAE rats as compared to control rats (0.66±0.17 in controls vs. 0.50±0.099 in EAE rats). However, a Student t-test showed there was no significant difference between the 2 groups (Fig. 5). A paired-pulse paradigm with 120-msec ISI shows facilitation of the second population response. Statistical analysis of the PPI in control (n=9) and in EAE (n=6) rats with the Student t-test showed that PPI was significantly decreased in the EAE group as compared to control group (1.33±0.50 in controls vs. 0.78±0.26 in EAE rats, P=0.024) (Fig. 6). Representative recordings of paired-pulse responses at 20- and 120-msec interpulse intervals in control and EAE rats are shown in Fig. 4.

Comparison of the paired-pulse index (PPI) in control (n=10) and experimental autoimmune encephalomyelitis (EAE) (n=8) affected rats at a 20-msec paired-pulse interval. The PPI was decreased in EAE rats compared to control rats. Statistical analysis with Student t-test showed there was no significant difference between the 2 groups (P=0.455).

Comparison of paired-pulse index (PPI) in control (n=9) and experimental autoimmune encephalomyelitis (EAE) (n=6) affected rats at a 120-msec paired-pulse interval. The PPI was decreased in EAE rats compared to control rats. Statistical analysis with Student t-test showed a significant difference between the 2 groups. *P=0.024.

Representative recordings of paired-pulse responses at the pyramidal cell layer in control and EAE rats. (A) Paired-pulse response at 20-msec, and (B) at 120-msec interpulse interval.
DISCUSSION
The study’s results indicate that activation of the peripheral immune system spread to the CNS and affected hippocampal neural transmission and neuroplasticity. Assessing spatial learning and memory in EAE rats is difficult since the disease can adversely affect motor performance. In this study, we used electrophysiology to study changes in hippocampal function during EAE for two reasons: (1) it is possible to assess hippocampal function without depending on animal locomotor activity, and (2) extensive synaptic changes in the hippocampus are believed to be responsible for deficits in hippocampal-dependent learning and memory. Although the effect of EAE on hippocampal synaptic transmission has been previously reported, this is the first time that the effect of EAE on synaptic transmission and LTP was investigated in vivo.
Our results show that EAE caused a decrease in PS amplitude. Consistent with our results, an in vitro study on EAE showed that synaptic transmission was decreased in the CA1 region of the hippocampus [10]. In contrast, Di Filippo et al. reported that central and peripheral inflammation do not change basal synaptic transmission [8,11]. in contrast than the above situation where there was no change in transmission, Centonze et al. [12] found that EAE induction in mice increased striatal synaptic transmission.
We have observed that EAE inhibits LTP induction in the hippocampus. Since LTP represents a synaptic model of learning and memory, its impairment could be associated with deficits in spatial learning and memory, which could explain the high prevalence of MS-associated cognitive dysfunction. In support of this data, other studies have shown that long-term plasticity was decreased in EAE animals as compared to controls [8,11]. Recently, it was reported that anti-NMDA glutamate receptor antibodies are present in MS patients [13,14]. Therefore, the failure of HFS to induce LTP in the hippocampus could be explained by the presence of these autoantibodies. In contrast to our results, Nistico et al. [15] showed that in vitro, HFS could induce LTP in the hippocampal CA1 region. Moreover, the LTP levels were significantly enhanced in EAE slices as compared to controls [15]. The disparity in the data in regards to synaptic transmission and LTP induction may be attributed to different recording sites (CA1 region vs. striatum) or recording methods (in vitro vs. in vivo).
PPI is a phenomenon observed at many chemical synapses and reflects changes in neurotransmitter release [16]. Pairedpulse index were usually recorded with interpulse intervals between 20 and 300 msec. An inhibitory postsynaptic potential (IPSP) mediated by the GABAA receptor and Cl-, reaches its peak within 10–50 msec and lasts about 100 msec [17]. As a result, we chose a 20-msec interpulse interval, which is selective for GABAA-mediated events [18]. Our results showed that EAE induction did not significantly alter PPI. Considering that a K+-mediated GABAB IPSP slowly reaches its peak within 13– 230 msec [18,19], we used a 120-msec interpulse interval to measure changes caused by EAE in GABAB-mediated events. We observed a significant reduction in the PPI of EAE animals as compared to controls, thus indicating that the efficacy of presynaptic GABAB receptors is increased in the hippocampal CA1 area of EAE rats. Presynaptic GABAB receptors are located at glutamatergic nerve terminals that regulate the release of glutamate from Schaffer collaterals to CA1 synapses [20,21]. Therefore, EAE might modulate suppression in synaptic transmission and synaptic plasticity by increasing the inhibitory effect of GABAB receptors on glutamate-mediated EPSPs.
In conclusion, the current study investigated the effect of EAE on synaptic transmission, LTP induction, and PPI in Wistar rats. The findings suggest that EAE might suppress synaptic transmission and LTP by increasing the inhibitory effect of GABAB receptors in glutamate-mediated EPSP. This mechanism may explain the detrimental effect of EAE on the learning abilities of rats.
Notes
Fund Support
Financial support for this study was provided by Deputy Vice-chancellor of research on Arak University of Medical Sciences Grant #1048.
Research Ethics
Ethical approval for the study was provided by the Arak University of Medical Sciences Research Ethics Committee # 92-159-11.
Conflict of Interest
No potential conflict of interest relevant to this article was reported.