INTRODUCTION
The urothelium has conventionally been regarded as a boundary between urine and the urinary tract. However, a number of studies have shown that it also possesses properties that are characteristic of organs capable of perceiving diverse signals from the urinary bladder [1]. Thus, the urothelium can function as a target organ for neurotransmitters (from the urinary bladder), release chemical mediators, as well as respond to external stimuli by expressing sensory molecules [2].
Meanwhile, several studies on the role of aquaporins (AQPs) in the urothelium or smooth muscles of the urinary bladder have illustrated the possible relation between AQPs and bladder dysfunction [3-6]. AQPs are transmembrane proteins that are involved in ion or water transport across the cell membranes [7]. They are expressed in cells like those of the kidney, which have a functional role in high rates of water transport. Interestingly, though, they are also frequently found in cells not involved in active water transport, such as the vascular endothelium and smooth muscle cells [7,8].
Flask-shaped caveolae are small invaginations of the cell membrane that are easily found in well-differentiated cells, such as those of the muscle. Caveolins (CAVs) are integral membrane proteins of the caveolae, and are essential for their formation. Moreover, these proteins have an important functional role in cell signaling, whereby they serve as scaffolds to foster the compartmentalization and integration of signals [9]. There are three isoforms of CAVs: CAV1, CAV2, and CAV3. Among them, CAV1 has been shown to play a role in the regulation of signal transduction pathways in a variety of cells [10]. Furthermore, Lai et al. [11] demonstrated that CAV1 knockout (KO) mice lack caveolae in the urinary bladder, and represent a number of urological disorders. In addition, CAV1 KO mice also exhibit decreased acetylcholine excretion and disrupted M3 muscarinic cholinergic activity of the bladder, leading to impaired smooth muscle contraction.
In our previous reports, we demonstrated altered expression of AQP1 in the urinary bladder of rats presenting detrusor overactivity [12-14]. Further, in a more recent study, we speculated about the relation between AQP1 and CAV1 in the urinary bladder, and found that they are significantly overexpressed upon detrusor overactivity (caused by bladder outlet obstruction [BOO]). Moreover, AQP1 and CAV1 were found to colocalize in the suburothelial microvasculature [12-14]. Based on earlier observations, we suggested that AQP1 and CAV1 might be closely associated with bladder signal activity that may, in turn indicate their role in detrusor overactivity. To date, however, no study has assessed the interrelationship between AQPs and CAVs in the urinary bladder, using CAV KO mice models. Thus, the purpose of this study is to investigate the expression of AQP1 in wild type and CAV1 KO mice, in order to determine their potential roles in the urinary bladder. We have specifically focused on the suburothelium, since this is the current target for research on bladder dysfunction.
MATERIALS AND METHODS
Animal Models
Young female C57BL/6J mice (8–10 weeks old; n=10), and CAV1 KO mice (backcrossed to C57BL/6J for 10 generations; n=10) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). The mice were inbred by homozygous mating. All mice were maintained in a pathogen-free animal center at the Clinical Vaccine Center of Chonnam National University (CNU IACUC-H-2009-80).
The mice were anesthetized with xylazine (2.2 mg/kg, intramuscularly) and zolazepam/tiletamine (4.4 mg/kg, intramuscularly). All mice procedures were performed according to the guidelines of the Animal Care and Use Committee of Chonnam National University. This study was permitted by the Ethics Committee of the Chonnam National University Medical School.
Western Blot
Tissues (n=10) were homogenized in ice-cold isolation solution with a Tissumizer homogenizer (Tekmar, Cincinnati, OH, USA) by a micro-saw tooth generator (five bursts, five strokes). Total protein was isolated from the homogenized tissues, and resolved by gel electrophoresis (12% sodium dodecyl sulfatepolyacrylamide; 50 خ¼g of protein). They were then transferred to polyvinylidene difluoride membranes (Amersham Pharmacia Biotech Inc., Piscataway, NJ, USA). Subsequently, the membranes were washed with Tris-buffered saline with Tween 20 buffer (10-mM Tris-HCl, pH 7.6; 150-mM NaCl; 0.05% Tween-20). They were then blocked with 5% nonfat dry milk, and incubated with the respective primary antibodies: rabbit polyclonal anti-AQP1 antibody (1:500; Santa Cruz Biotechnology Inc., Dallas, TX, USA), mouse monoclonal anti-CAV1 (1:1,000; BD Biosciences, San Jose, CA, USA), and rabbit polyclonal antiglyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (1:4,000; Cell Signaling Technology, Danvers, MA, USA). Horseradish peroxidase-conjugated antirabbit antibody for AQP1 (1:2,000; Santa Cruz Biotechnology Inc.) and GAPDH (1:4,000; Sigma, St. Louis, MO, USA) were the secondary antibodies used. Protein bands were detected by WESTZOL plus Western Blot Detection System (iNtRON Biotechnology Inc., Seongnam, Korea), and visualized with LAS-3000 (Fujifilm Life-science, Tokyo, Japan). Densitometric scanning was conducted by the Multi gauge V3.0 (Fujifilm Life-science) analysis software and chemiluminescence system.
Immunofluorescence Staining
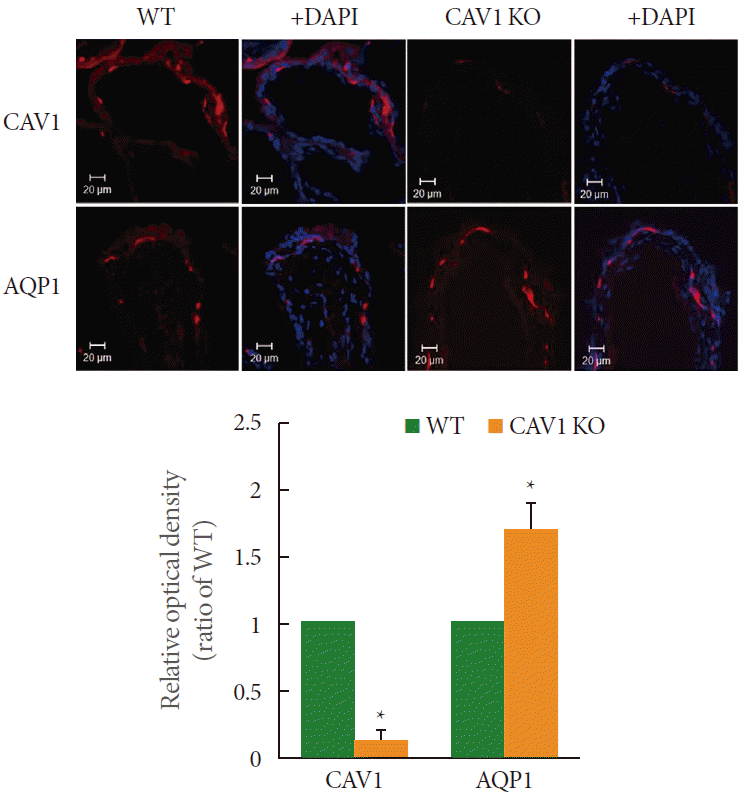
The tissue sections (10 for each tissue) were washed in phosphate buffered saline, and incubated with normal chicken serum (3%, 30 minutes, room temperature) to prevent nonspecific binding. They were then incubated with antibodies against AQP1 (1:100; Santa Cruz Biotechnology Inc.) and CAV1 (1:100; BD Biosciences) for 12–14 hours at 4آ°C. Immunoreactivity for CAV1 was detected using Alexa Fluor 594 chicken antimouse IgG (H+L), and for AQP1 by Alexa Fluor 594 chicken antirabbit IgG (H+L) (1:100; Molecular Probes Inc., Eugene, OR, USA). DAPI (4آ´-6-diamidino-2-phenylindole) (Vector Laboratories, Burlingame, CA, USA) was used for tissue mounting, to stain the nucleus. For negative controls, tissues were similarly processed, but without AQP1 and CAV1 antibodies. Tissues were visualized with a confocal microscope (LSM 510, Carl Zeiss Korea, Seoul, Korea) with wavelengths appropriate for Alexa Fluor (594 nm) and Alexa Fluor (405 nm). Densitometric analysis was performed with Studio Star Scanner using the NIH Image V1-57 software.
RESULTS
There was no difference in body weights (24.2آ±0.4 g vs. 23.9آ±0.5 g) and bladder weights (44.8آ±1.2 mg vs. 45.1آ±0.8 mg) between the two groups (control vs. CAV1 KO).
Expression of AQP1 in CAV1 KO Mice, as Detected by Western Blotting
Western blot analysis revealed bands for AQP1 and CAV1 at 28 kDa and 22 kDa, respectively (Fig. 2). Both the proteins were detected in the wild type group, whereas CAV1 was not detected in the CAV1 KO mice. Moreover, the expression of AQP1 was significantly increased in the CAV1 KO mice (P<0.05) (Fig. 2), in corroboration with the immunofluorescence data.
DISCUSSION
The present study demonstrated increased expression of AQP1 in the suburothelial microvasculature, in CAV1 KO mice urinary bladder. The immunohistochemical study showed that AQP1 and CAV1 in the urinary bladder colocalized in the arterioles, venules and capillaries of the suburothelial layer. Collectively, these results provide evidence that mice lacking CAV1 significantly overexpress AQP1 at the urothelium, which is actively involved in afferent signal transduction. Consequently, AQP1 and CAV1 might be closely associated, and may play a role in bladder dysfunction. To the best of our knowledge, this is the first in vivo study that demonstrates the possible manifestation of AQP1/CAV1-mediated suburothelial cell signaling in bladder dysfunction.
Various studies have implicated that the urothelium can mediate solute and water transport under specific circumstances [15,16]. The epithelial sodium channel (ENaC) is well known to be responsible for fluid and salt transport across epithelial cell membranes. Investigations have revealed that ENaC shows significantly greater expression in BOO patients with clinical symptoms of bladder dysfunction than in the controls. Clinically, this correlated with the storage symptom score of the patients [17]. However, studies on the expression of AQPs in the urinary bladder are limited. In our previous report, detrusor overactivity caused by BOO led to a significant increase in the expression of AQP2–3, and nitric oxide synthase (endothelial nitric oxide synthase, neuronal nitric oxide synthase) in rat urinary bladder. In the study, we suggested that the AQPs and nitric oxide synthase isoforms might have a specific role in bladder dysfunction that is related with bladder change following BOO [12].
The CAV1 KO mouse has been proposed as a novel animal model to study bladder impairment associated with primary hypocontraction of urinary bladder and detrusor overactivity [11,18]. Studies on CAV1 KO mice have established that loss of CAV1 is associated with disrupted muscarinic cholinergic activity. This induces impaired smooth muscle contraction in the urinary bladder upon stimulation with carbachol [19,20]. Further, in the organ bath study using bladder strips of CAV1 KO mice (produced by genetic ablation of caveolae), a 70% decrease in acetylcholine release from bladder nerve terminals was observed [11]. However, the functional importance of these organelles in relation to the micturition response in the urinary bladder has not been entirely delineated.
Intriguingly, until date, there are no studies either on the relation between AQPs and CAVs in the suburothelium (with regard to bladder dysfunction), or the alteration of their expressions in CAV1 KO mice. Gao et al. [21], however, had revealed a functional interaction between AQP1 and CAV1 in the change of microvasculature and regulation of alveolar epithelial permeability in rat lung. They reported that CAV1 knock down by small interfering RNA causes increased microvascular and epithelial permeability as well as pulmonary edema. They also proposed that CAV1 plays an important role in the regulation of pulmonary permeability by modifying AQP1 [21].
It is important, however, to note the limitations of this study: Our hypothesis warrants in-depth studies showing (1) the physiological alteration of the urinary bladder. to confirm the CAV1/AQP1 association at the suburothelium; (2) the functional signal change(s), or change in solute or water movement; and (3) specific functional changes in bladder activities. Deeper understanding of the AQP1/CAV1 relation is likely to explain the cellular mechanisms of bladder dysfunction, and may suggest potential molecular targets for its pharmacological treatment.
In conclusion, our results imply that lack of CAV1 may result in significant upregulation of AQP1, providing possible evidence that these proteins are closely associated in the urothelium. This is probably via modification of cellular transmission and signaling pathway(s) specific to the suburothelial tissue. The possible interrelation between AQP1 and CAV1 also stems from the fact that they colocalized at the endothelium of the microvasculature, which is important in terms of blood supply to the urothelial tissues.