Pathophysiology of Overactive Bladder and Pharmacologic Treatments Including β3-Adrenoceptor Agonists -Basic Research Perspectives-
Article information

Abstract
Overactive bladder (OAB) is a symptom-based syndrome defined by urinary urgency, frequency, and nocturia with or without urge incontinence. The causative pathology is diverse; including bladder outlet obstruction (BOO), bladder ischemia, aging, metabolic syndrome, psychological stress, affective disorder, urinary microbiome, localized and systemic inflammatory responses, etc. Several hypotheses have been suggested as mechanisms of OAB generation; among them, neurogenic, myogenic, and urothelial mechanisms are well-known hypotheses. Also, a series of local signals called autonomous myogenic contraction, micromotion, or afferent noises, which can occur during bladder filling, may be induced by the leak of acetylcholine (ACh) or urothelial release of adenosine triphosphate (ATP). They can be transmitted to the central nervous system through afferent fibers to trigger coordinated urgency-related detrusor contractions. Antimuscarinics, commonly known to induce smooth muscle relaxation by competitive blockage of muscarinic receptors in the parasympathetic postganglionic nerve, have a minimal effect on detrusor contraction within therapeutic doses. In fact, they have a predominant role in preventing signals in the afferent nerve transmission process. β3-adrenergic receptor (AR) agonists inhibit afferent signals by predominant inhibition of mechanosensitive Aδ-fibers in the normal bladder. However, in pathologic conditions such as spinal cord injury, it seems to inhibit capsaicin-sensitive C-fibers. Particularly, mirabegron, a β3-agonist, prevents ACh release in the BOO-induced detrusor overactivity model by parasympathetic prejunctional mechanisms. A recent study also revealed that vibegron may have 2 mechanisms of action: inhibition of ACh from cholinergic efferent nerves in the detrusor and afferent inhibition via urothelial β3-AR.
INTRODUCTION
Overactive bladder (OAB) is a very common urological disease worldwide, and the prevalence rate is estimated to be approximately 11.8%–16.6% [1-3]. OAB is a symptom-based syndrome defined by urgency, frequency and nocturia with or without urge incontinence [4]. It may be overlooked since it is not a life-threatening disease like cancer; however, as symptoms worsen, it may greatly affect daily life and reduce the quality of life. It may cause depression and anxiety, and it has been reported that nocturia in elderly OAB patients is closely associated with fractures, sleep disorders, and an increased prevalence of cardiovascular disease [5, 6].
Because OAB is a symptom-based syndrome, the causes are multifactorial. Therefore, there also be various phenotypes. Detrusor overactivity (DO), often confused with OAB, is strictly a urodynamic finding characterized by involuntary detrusor contractions during the filling phase, which may be spontaneous or provoked [4]. While the majority of men with DO in urodynamic findings represented urgency (90%), not all patients with urgency have DO (69% in men vs. 44% in women) [7]. Many studies to reveal pathological mechanisms are conducted using animal experiments. Because it is impossible to objectively measure the urgency symptom in noncommunicative animals, most OAB animal models are based on those with DO [8], in which involuntary detrusor contraction can be measured on the cystometrogram (CMG). Therefore, unfortunately, experimental models cannot represent all clinical pathologies perfectly. Nevertheless, over past few decades, we have been able to detect the pathophysiological mechanisms underlying OAB, which had previously been regarded as idiopathic.
There has also been a lot of progress in the therapeutic fields of OAB. In addition to antimuscarinics, β3-adrenergic receptor (β3-AR) agonists and botulinum toxin A have expanded the treatment options. Invasive procedures such as sacral neuromodulation and peripheral tibial nerve stimulation have also become a part of OAB treatments. Particularly, β3-AR agonists play an inhibitory role in the afferent signaling pathway of the bladder and have the advantage of minimal or no decrease in detrusor contractility compared to antimuscarinics. Recently, it has been suggested that continuous treatment with these drugs may inhibit neural remodeling in the central nervous system (CNS) in an OAB animal model [9]. In this article, we reviewed the OAB pathophysiology, the pharmacological treatment options commonly used in clinical practice, and its mechanisms of action. Additionally, the action mechanisms of β3-AR were reviewed under various pathologic conditions.
PATHOPHYSIOLOGY OF OAB
OAB pathophysiology has mostly been approached from 3 major perspectives. Neurogenic theory can be explained by degenerative changes or damages in the nerve pathway involved in the micturition reflex [10]. Another perspective, the myogenic hypothesis, is that the changes in the muscle cells of the bladder may lead to bladder overactivity [11]. More recently, the urothelial role in the changes of bladder afferent signaling pathway, which result in OAB, has been proposed [12]. Other researchers have also focused on continuous stimulation transmitted from the bladder to the CNS, regardless of whether the source is nerve, muscle, or urothelium. These stimuli, which fire spontaneously like a pacemaker, are also referred to as micromotion, spontaneous contraction, or afferent noises and are attracting attention as one of the causes of bladder overactivity [13, 14].
Neurogenic Hypothesis
Neurogenic factors have 2 main causes. One is that the inhibitory system, which controls the micturition reflex, is damaged and does not work properly, and the other is that the micturition reflex is enhanced [10]. Reduced suprapontine inhibition due to brain injury [15, 16], damaged inhibitory axonal transmission from the CNS due to spinal cord injury (SCI) [10], and loss of peripheral inhibition are included in the damaged inhibitory system of the micturition reflex. Causes of enhancement of the micturition reflex include abnormally increased afferent activities from the bladder [14, 17, 18] and increased excitatory neurotransmission from the CNS by neural remodeling [10] (Fig. 1).
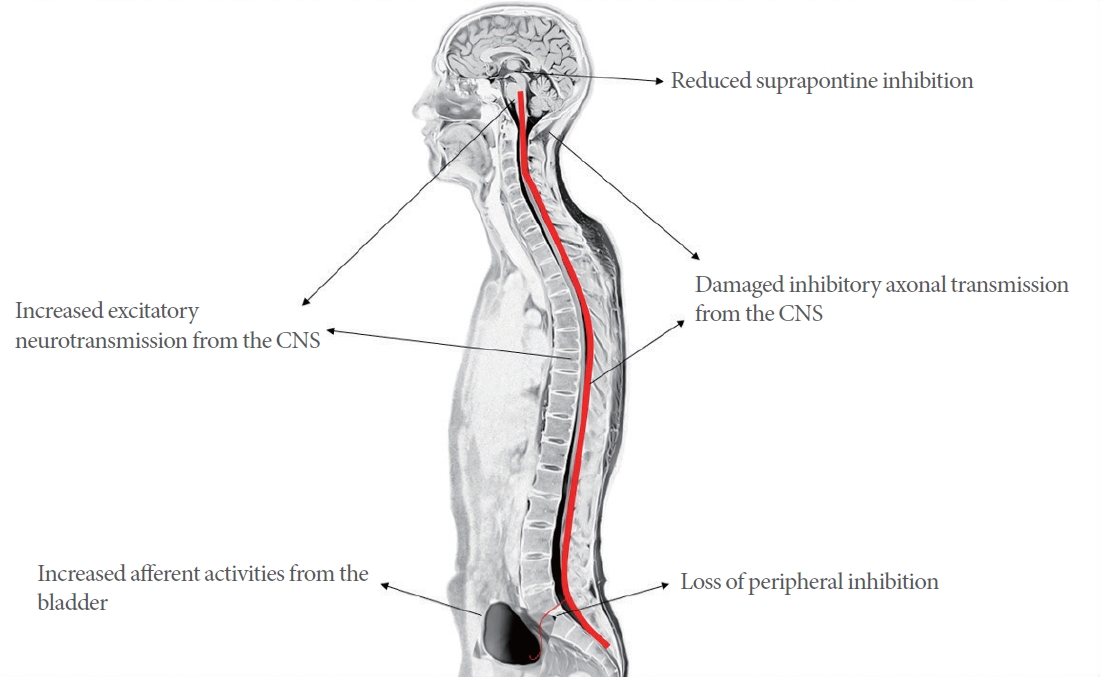
Pathophysiology of neurogenic etiologies in overactive bladder. CNS, central nervous system.
Diseases, which affect the CNS such as stroke, Parkinson disease, multiple sclerosis, and SCI, are often accompanied by OAB symptoms [17]. The cerebral cortex acts as a tonic inhibition system, which suppresses parasympathetic excitatory outflow from the bladder during the storage phase. If this region is damaged, suprapontine inhibition is reduced, resulting in DO [19]. Glutamatergic excitatory transmission is known to be associated with bladder overactivity. In rats with decreased bladder capacity due to cerebral infarction, administration of N-methyl-D-aspartate glutamatergic antagonist counteracted this effect [20]. In a Parkinson disease animal model using N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a neurotoxin which can destroy dopamine neurons, the D-1 dopaminergic receptors inhibited bladder overactivity and D-2 dopaminergic receptors were found to facilitate micturition [21, 22]. Urgency induced by large bladder volume without DO showed an exaggeration of cortical responses, for which the anterior cingulate gyrus is found to be the central region. However, in cases accompanied with DO, prefrontal deactivation was a distinct finding [23, 24]. The thalamus also seems to modulate the lower urinary tract function, and thalamic deep brain stimulation induced the urge to urinate earlier and decreased bladder capacity [25]. Recent brain imaging studies have also shown that bladder control is associated with an extensive network of brain regions. Thus, it is thought that dysfunction in various areas of the CNS may cause different phenotypes of OAB [23].
In periphery, bladder afferents are located in the suburothelial layer and interact and participate in signal transmission through numerous excitatory and inhibitory transmitters released from the urothelium. Dysregulation of bladder afferent activity results in changes in micturition signals in the efferent pathway, resulting in detrusor dysfunction [26]. Although the purinergic component involved in nerve-mediated contraction was not identified in the normal bladder specimens, approximately 50% of the purinergic component was identified in OAB specimens [27]. Abnormally activated purinergic transmission may be related to OAB symptoms. Some researchers have also paid attention to increased afferent activity mechanisms. Oxybutynin, an antimuscarinic agent, appears to inhibit the afferent part of the micturition reflex by not only relaxing the detrusor muscle but also affecting the bladder’s sensory nerve function [28]. In rats, CL316,243, a β3-AR agonist, inhibited Aδ-fibers but not mechanosensitive C-fibers. However, CL316,243 could also suppress PGE2-induced C-fiber hyperactivity [29].
The detrusor-to-detrusor reflex is mediated through 2 peripheral afferents (Aδ- and C-fiber afferents) [30, 31]. The C-fiber afferent-evoked reflex in spinal intact (SI) animals does not respond to bladder distention and, under normal conditions, the reflex through the C-fiber is weak and only partially responsible [32]. Electrophysiological studies have shown that the conduction delay of animals with SCI in the micturition reflex is relatively shorter than that of SI animals [31, 33, 34], indicating that the afferent limb of the micturition reflex is composed of unmyelinated C-fibers after SCI [30]. Capsaicin, a neurotoxin known to desensitize C-fiber afferents [35], failed to block the Aδ-fiber-evoked bladder reflex in SI animals, but could block the C-fiber-evoked bladder reflex in SCI animals [30, 31]. Additionally, in SCI patients with DO and autonomic dysreflexia, intravesical administration of capsaicin induces increased bladder capacity and decreased contraction pressure, autonomic dysreflexia, and urge urinary incontinence (UUI) [36, 37]. These results indicate that axonal damage to the spinal cord reorganizes the micturition reflex pathway, resulting in hyperexcitability of the C-fiber afferents [10, 26]. SCI also changed the muscarinic presynaptic modulatory mechanism in the cholinergic terminal of the bladder [38]. These changes enhance parasympathetic signaling and seem to be related to DO.
Myogenic Hypothesis
Predisposing factors such as partial denervation and bladder ischemia may alter the properties of the detrusor smooth muscle [39-41], and DO may result from the histologic changes in the detrusor [42], which may result in spontaneous, autonomous cellular activity mediated by extracellular Ca2+ influx and intracellular Ca2+ release [43]. It has been reported that this local contraction and micromotion, which begin at some parts of the bladder, spread throughout the bladder wall, resulting in coordinated myogenic contraction [39, 44, 45]. Coordinated myogenic contraction and increased intravesical pressure can then generate urgency by transferring afferent signals to the CNS.
Urothelial Hypothesis
In the past, the urothelium was thought to be a simple barrier that isolate the bladder from the urine. However, more recently, it has been discovered that the urothelium is an important sensory organ that senses and communicates thermal, mechanical, and chemical stimuli beyond the passive barrier and plays an important immunological role in the pathogenesis of diseases such as OAB or interstitial cystitis/bladder pain syndrome (IC/BPS) [46]. Spontaneous detrusor contraction modulated by bladder mucosa showed low amplitude and high frequency activities in SI rats, but became high amplitude and lower frequency in SCI rats. Partial removal of the mucosa reduced the amplitude of spontaneous contraction and the response to bladder stimulation with stretch or chemical stimuli. In tissues where mucosa was removed, enhanced spontaneous activity was eliminated. Under manipulated conditions of suppressed smooth muscle signals, spontaneous contraction in the pathologic bladder was driven by mucosa. These results suggest the possibility that spontaneous contraction originated from the urothelium [47].
Urothelial cells can be targets of transmitters secreted by nerves or other types of cells. It may also be activated through autocrine or paracrine mechanisms. Urothelium and suburothelium contain afferent nerves and receptors. During the storage phase, compounds produced and secreted here activate or inhibit the afferent pathway. Excitatory and inhibitory neurotransmitters such as acetylcholine (ACh), adenosine triphosphate (ATP), and nitric oxide (NO) secreted from the urothelium are involved in this mechanism [46].
Since much evidence suggested that the abnormal sensory function seen in OAB may be due to increased activity of bladder afferents, urothelial and suburothelial dysfunction has received attention. Alterations in the function of the urothelial receptor, the release of neurotransmitters, the sensitivity of interstitial cells in the suburothelial layer, and their coupling may induce involuntary bladder contractions [48, 49]. Thus, spontaneous contractions from mucosa have been suggested as a possible cause of urgency.
The receptors of nerve growth factor (NGF) are abundantly expressed in the urothelium. NGF is increased in the bladder and urine of patients with bladder outlet obstruction (BOO), diabetic cystopathy, neurogenic DO, IC/BPS, and other types of storage lower urinary tract dysfunction (LUTD) [48, 49]. Transient receptor protein cation channel subfamily V member 1 (TRPV1) is a nociceptor known to transmit and modulate pain in response to temperature, acidity, capsaicin, etc. NGF stimulates the proliferation and survival of target neurons [50], and bladder NGF also lowers the threshold of TRPV1 [51]. Excessive NGF expression is involved in OAB pathogenesis by influencing bladder dysfunction through these mechanisms [52]. Some studies have found that unidentified inhibitory substances other than NO, cyclooxygenase, catecholamine, adenosine, and GABA (γ-Aminobutyric acid) are released from the urothelium upon stimulation of muscarinic receptors [53, 54]. Additional future research is necessary to elucidate the relationship between various urothelially released these substances and the etiology of OAB.
Autonomous Activities (Afferent Noises)
Pre- and postganglionic ACh from the parasympathetic nerve may be leaked from the parasympathetic nerves by bladder stretching, even during the normal storage phase. This leakage of ACh is increased in DO. In this process, the sensitivity of detrusor muscle cells to neurotransmitters increases, resulting in local contractions (micromotion) of the detrusor bundle. Although this local contraction is not coordinated and cannot increase intravesical pressure in all cases, it may generate an afferent signal, which triggers the micturition through the pontine micturition center, may eventually coordinate bladder contractions [13].
Previous studies have reported that bladder smooth muscle autonomous activity, nonmicturition contractions, and phasic sensory discharge are all features found during normal bladder filling. The afferent discharge (afferent noises) associated with this response may be induced by stretch, noxious stimuli, and chemicals released from the urothelium or mediated by the motor or sensory system [55]. In addition, several other systems appear to be involved in afferent noises. When the afferent information from the bladder is vast, the CNS is overflowing with such afferent noises from many other sources. However, it is unclear how and when the CNS selects the afferent noses necessary to trigger the micturition reflex or perceive sensation although the findings appear to be somewhat different from normal micturition mechanisms [55].
Afferent information transmits useful information to the CNS during micturition and storage. When afferent noises reach a certain intensity, the body seems to interpret them as various stages of bladder filling, including urgency or pain. Only some part of the afferent noise is used to create sensation, and the others probably contribute to bladder filling, sphincter control, and coordination of micturition reflexes [55]. Hyperexcitability of the nervous system, CNS and peripheral nervous system due to pathological conditions and defects in inhibitory mechanisms seem to alter autonomous bladder activity, resulting in DO [14, 56]. The findings of increased rhythmic activity in the bladder in the BOO rat model [57] and increased micromotion of the bladder in women with chronic pelvic pain [58] are supporting evidence. It is similar to the myogenic hypothesis that these autonomous activities occur within the bladder wall component, and there are some overlaps with the neurogenic hypothesis, in which the CNS can influence them.
PATHOLOGICAL CONDITIONS UNDERLYING OAB
BOO and Bladder Ischemia
DO is highly prevalent (approximately 52%–60%) in patients with BOO due to BPH [59]. In a study for preoperative patients, the prevalence rate of OAB without or with BOO was reported to be 25% or 62%, respectively, showing that BOO is closely related to the occurrence of OAB [60, 61]. Chronic BOO causes pathophysiological alterations in all layers of the bladder wall, including detrusor hypertrophy, fibrosis, urothelial dysfunction, and functional denervation [62, 63]. In a BOO model using rodents, partial BOO resulted in bladder dysfunction accompanied by bladder hypertrophy and fibrosis [64]. Bladder distention due to inadequate emptying may induce bladder ischemia, and repeated reperfusion increases the release of free radicals and cytokines and causes inflammation, resulting in reperfusion damage in tissues [65]. In terms of ischemia, hypoxia-inducible factor (HIF) and proinflammatory cytokines, including interleukin-1β (IL-1β) and transforming growth factor-β (TGF-β), increase [66]. In a recent study, an increase in the expression of toll-like receptor 4 (TLR-4) and TLR-9, which are related to inflammation induction, was reported in the urothelium of the BOO animal model. Antagonists of these receptors inhibited BOO-induced inflammatory responses [65]. In the urothelium of the BOO bladder, nucleotide-binding oligomerization domain-like receptors family pyrin domain containing 3 (NLRP3) inflammasome was also activated, which promotes fibrosis and detrusor denervation [67]. Inhibitors of NLRP3 prevented these alterations and preserved bladder function in partial BOO mice [68]. Bladder overactivity caused by BOO also appears to be associated with changes in NGF, potassium channel subfamily K member 2 (KCNK2, also known as TREK-1), other potassium channels, muscarinic- and purinergic receptors. In the BOO model using rats, NGF was involved in the interaction between target organs and nerves, resulting in neuroplasticity [69]. TRPV1 was expressed not only in the afferent nerve of the bladder but also in urothelial cells [70]. Changes in the expression of NGF and TRPV1 in the bladder seem to affect sensory signaling and result in neuroplasticity in the CNS. This neural remodeling, central sensitization, may be a factor in OAB symptoms persisting even after BOO is resolved [9, 71, 72]. TREK-1 is part of a subfamily of mechano-gated potassium channels and stabilizes the membrane potential of detrusor myocytes during bladder filling. This action relaxes the bladder wall to maintain low pressure while the bladder is filled with urine [73]. In the BOO mice model, protein expression and immunoreactivity of the TREK-1 channel were significantly reduced in detrusor smooth muscle. L-methioninol, a TREK-1 channel blocker, significantly increased premature contraction during the filing phase in sham-operated mice although it did not significantly affect BOO mice with DO. These results show that a decrease in the TREK-1 channel is associated with the OAB-like condition in the BOO mouse model [74]. Activation of certain types of potassium channels stabilizes membrane potentials and reduces the excitability of nerves and muscle cells. Several studies have suggested the relevance of potassium channels in OAB [75-78]. For example, in the rat bladder at 6 weeks after BOO, the expression of the β1-subunit of large conductance calcium-activated potassium (BK) channels and small conductance calcium-activated potassium channel 3 (SK3 channel) were significantly increased, whereas the BK channel β4-subunit expression was decreased as the severity of BOO worsens. These results indicate that, in the early stages of BOO, BK and SK channels may increase to suppress bladder contraction as a compensatory mechanism for BOO-induced OAB; however, during OAB progression, this compensatory mechanism seems to fail to work [79]. Also, the immunoreactivity of M2-, M3-, and P2X3 receptors was simultaneously increased in the urothelium of BOO rats [80]. Muscarinic receptors are expressed in the urothelium as well as the detrusor muscle and are involved in the OAB pathogenesis [81, 82]. Purinergic P2X3 receptors located at the suburothelial sensory afferents are activated by ATP released by urothelial stretching, and intravesical instillation of ATP could induce bladder overactivity in rats [83, 84]. Thus, ATP and P2X3 also seem to be involved in urothelial signaling and OAB pathophysiology.
In addition, BOO and arterial occlusive disease are wellknown etiologies of bladder ischemia and have been commonly used as a bladder ischemia animal model. In BOO patients, an increase in outlet resistance during voiding may cause compensatory bladder hypertrophy, resulting in a perfusion deficit of tissue [85]. The average bladder wall thickness, particularly in trigone, was closely related to urgency in OAB women [86].
A study of the ischemic rat model using iliac artery injury showed DO at 8 weeks from the injury. However, detrusor underactivity was found at 16 weeks from the injury, indicating that DO can emerge by compensatory mechanisms in the early period of bladder ischemia. Accordingly, the expression of the M3 muscarinic receptor was increased at 8 weeks from injury but decreased at 16 weeks. Also, the histological findings showed degeneration of both muscles and nerves over time [87]. Ischemic stress in the bladder can be detected by cellular stress sensors such as 5’ adenosine monophosphate-activated protein kinase, apoptosis signal-regulating kinase 1, and caspase-3 [88]. In bladder ischemia-induced DO animal models, factors such as HIF, TGF-β, vascular endothelial growth factor, and NGF were increased, which may play an important role in the OAB pathophysiology related to ischemia [89].
Obesity and Metabolic Syndrome
Metabolic syndrome has a prevalence of approximately 23% in adults and consists of risk factors such as cardiovascular disease, diabetes, insulin resistance, central obesity, dyslipidemia, and hypertension. There was no gender difference in the prevalence of OAB among men and women with metabolic syndrome [90-93]. In men with metabolic syndrome, incidences such as nocturia, incomplete emptying, weak urinary flow, and hesitancy were increased [94]. Also, the impact of diabetes mellitus (DM) on lower urinary tract symptoms (LUTS) is multifactorial. DM may cause dysfunction of the detrusor smooth muscle, urothelium, and nerves [95]. In a study of 1,359 DM patients, 22.5% had OAB, and in those over 60 years of age, the distribution was slightly greater in men than in women (24.8% vs. 20.1%). Regarding BPH, cases with DM tended to have larger prostates than those without DM [96, 97]. Diuresis and metabolic effects due to DM resulted in detrusor hypertrophy and changes in mechanical properties, which decreased bladder voiding efficiency. In the early stages of DM neuropathy, bladder overactivity was observed. In streptozotocin-induced DM rats, M2- and M3 receptor expression increased in the urothelium and bladder muscle at 2 weeks [98, 99]. In another study, an underactivity was shown at 12 weeks after DM induction, accompanied by increased urine NGF, EP1, and EP3 (E-series prostaglandin receptors), but by decreased bladder NGF and urine PGE2 [100]. In an animal model of metabolic syndrome with long-term fructose feeding, upregulation of M2- and M3 receptors were implicated in DO, and metabolic perturbations due to fructose intake resulted in increased proinflammatory cytokines in detrusor muscles, increased oxidative stress, mitochondrial dysfunction, and increased apoptosis. Additionally, detrusor hypertrophy seemed to contribute to DO [101, 102]. Detrusor hypertrophy observed in metabolic syndrome or diabetic animal models was accompanied by decreased functional bladder capacity and increased urinary frequency. Hypertrophy typically presents with reduced compliance, high intravesical pressure, and DO, which may lead to reperfusion injury [103]. Furthermore, mitochondria can provide high energy consumption in the early stages of bladder hypertrophy, but long-term and excessive energy consumption depletes and deforms mitochondria, resulting in mitochondrial damage [104, 105].
Hyperlipidemia may be associated with OAB women, according to some clinical studies [106]. Experimental studies using rats demonstrated that a decrease in detrusor contractility in the hypertension and hyperlipidemia model was related to a decrease in Rho kinase and protein kinase activity [107]. A study using chronic hyperlipidemic rabbits reported reduced bladder capacity, DO, and nerve degeneration as a result [108]. In a bladder ischemia model using iliac artery injury, vascular injury accompanied by high-fat diet-induced hypercholesterolemia produced more severe ischemia than in vascular injury alone, and the mechanisms of this pathologic progress may be closely related to the occurrence of OAB [109]. Obesity, either alone or in combination with DM, was strongly associated with the development of OAB and LUTS, including SUI in women [110]. Hypertension, hyperinsulinemia, and obesity are also associated with autonomic hyperactivity, which can cause bladder dysfunction and LUTS [111]. Based on various research results, as the mechanism of OAB caused by metabolic syndrome, it is assumed that increased metabolic loads stimulate bladder sensory afferents, and various other factors such as oxidative stress, systemic inflammation, and insulin resistance promote chronic pelvic ischemia and urothelial dysfunction.
Psychological Stress and Affective Disorder
OAB patients are vulnerable to depression and anxiety because of their bothersome symptoms. Conversely, psychological stress and affective disorders may be the risk factors for developing or worsening OAB [112, 113]. There were alterations in the micturition pathway of CNS and local changes in bladder function during the investigation using an animal model of stress-induced bladder dysfunction. These local changes in the bladder included detrusor hypertrophy, increased bladder contractile response, and afferent hypersensitivity [114-116]. The bidirectional relationship between affective disorder/psychological stress and OAB may exist. Corticotrophin-releasing factor (CRF), which is commonly involved in mechanisms of both pathologies, provides with supporting evidence for this relationship [117].
The limbic-hypothalamic-pituitary-adrenal axis is important in behavioral, physiological, and molecular responses to stress conditions [118-120]. In particular, the paraventricular nucleus (PVN) of the hypothalamus produces and releases CRF and projects it to other sites in the body [118, 121, 122]. CRF is known to mediate stress and visceral hyperalgesia, etc. [112, 123, 124]. Different brain regions, such as the prefrontal cortex, amygdala, and hippocampus, as well as PVN, may be involved in stress responses [122]. Recently, it has been reported that stress-induced molecules such as bombesin, angiotensin II, nicotinic ACh receptor, NO, and hydrogen sulfide (H2S) stimulate various cerebral regions and affect the micturition circuit [125].
Receptors for CRF were located throughout the central micturition pathway and in the periphery of the urothelium, where they increase ATP release and contribute to the enhancement of pelvic sensory hypersensitivity [126]. Additionally, catecholamine from the adrenal medulla can trigger the release of cytokines throughout the nervous system [116]. CRF receptors are also located on inflammatory cells involved in innate immunity. Thus, stress-induced OAB may also be accompanied by an inflammatory response. An increase in CRF due to stress causes the release of cytokines from activated immune cells through CRF and releases cytokines through catecholamine. Therefore, it seems that activation of this pathway by stress may result in an inflammatory response in the CNS and the bladder and affect the lower urinary tract function through cytokine release. A study targeting volunteers reported that stress increases the proinflammatory cytokines IL-1β, IL-6, IL-10, and tumor necrosis factor-α in plasma [127]. Animal experiments using a CRF receptor 1 antagonist showed that depression-induced OAB was improved as serum CRF decreased. A decreased serotonin in the CNS also resulted in urinary frequency and DO [128] whereas frequency and urgency were improved in OAB patients treated with duloxetine, a norepinephrine serotonin reuptake inhibitor [129].
Central sensitization is a condition in which nociceptive neurons in the CNS respond excessively to stimulation with a threshold level below normal through the afferent pathway. It has also been suggested as another pathophysiologic cofactor of OAB and affective disorder [130]. Transient receptor potential (TRP) channels play an important role in the central sensitization process, and the dysfunction of TRP channels is considered important in the comorbidities of affective disorders and OAB [72, 130, 131].
Urinary Microbiome
Until recently, urine was considered sterile, and bacterial growth was abnormal as found in urinary tract infection (UTI). Additionally, the criteria for diagnosing OAB include the absence of infection. However, with the advancement of bacterial culture technology, what we knew was no longer true. Although the mechanism and causal relationship are still unclear, it seems likely that the microorganisms detected in urine are not abnormal findings anymore, and the microbiome may be involved in the mechanism of OAB development [132]. In one study, in which antibiotics were administered to OAB patients who tested negative for the conventional culture technique, symptoms improved in half of the patients [133]. In addition, using a new culture technology, approximately 39% of refractory OAB patients who had not been able to diagnose UTIs with existing culture technology had bacterial infections [134]. In UUI patients, bacterial DNA and higher bacterial load were found more frequently, and the diversity of the urinary microbiome was reduced [135-137]. Strains such as Lactobacillus crispatus found in healthy female bladders represented lower bacterial loads in patients with UUI [135, 136]. Lactobacillus seems to inhibit the growth of virulent bacteria in the same environment where they grow due to its acid-producing properties. Although intravaginal administration of Lactobacillus significantly reduced recurrent UTI, there is still a lack of evidence regarding the role of Lactobacillus in OAB [135]. Some studies have reported that the urine microbiome had a significant impact on OAB treatment results, and in particular, baseline characteristics with fewer bacteria and communities with less diversity responded better to the treatment.
In addition, recent studies have also shown that abnormal urinary microbiomes with less diversity were associated with higher levels of depression and anxiety [137], suggesting that the urinary microbiome can communicate with the brain. In other words, the brain-bladder axis driven by the microbiome may exist like the brain-gut axis, in which the gut microbiome can affect CNS directly through the systemic pathway across the blood-brain barrier (BBB) using metabolites such as short-chain fatty acids and may affect neuroglial interactions [138, 139]. This continuous interference creates neural remodeling in the CNS, and through bidirectional interference, it can also affect peripheral organs. If the brain-bladder-microbiome axis exists, it is assumed that bladder function will be affected along with CNS changes by the same mechanism. Like the mechanisms of psychological stress mentioned above, a top-down pathway from the brain may also cause LUTS. Local and systemic immune responses, including inflammation, can also affect the CNS directly and indirectly [140, 141]. Thus, there is the possibility of multidirectional communication between the bladder, CNS, and nervous and circulatory systems, which could help understand the mechanism of OAB and establish treatment strategies.
Inflammation
Inflammation is not a common feature of OAB. However, Tyagi et al. [142] reported that several markers related to inflammation were increased in the urine of OAB patients. In another study, bladder biopsies of patients with neurogenic DO showed severe and inflammatory infiltration in 24% and 74%, respectively [143]. A previous study also reported that pyuria was present in one-third of OAB patients, and a strong correlation between OAB severity and immune response was suggested by another report [144]. It has also been suggested that a relationship between inflammation and OAB, in which proinflammatory cytokines such as pyuria and IL-6 were significantly more evident in OAB patients than controls [145]. Although, based on these results alone, it is not easy to conclude whether OAB patients are susceptible to inflammation or whether inflammation is a preceding cause of OAB, several experimental studies suggest the hypothesis that inflammation may be one of the causes of OAB. For example, as previously mentioned, bladder ischemia may induce an inflammatory response, which produces neural remodeling of bladder afferent pathways. The metabolism of mucosa was 3 times higher than that of smooth muscle, and perfusion changes were greater in mucosa than in muscle [146, 147]. In addition, inflammatory cell infiltration was mainly observed in the suburothelial layer [143]. Based on these findings, mucosal membranes appear to be structurally vulnerable to ischemic damages. Moreover, as urothelial afferent nerves exist in the mucosa, several mediators within the mucosa affect detrusor contraction and afferent nerve activity. Alterations in the urothelial afferent signaling pathway due to inflammation are thought to be one of the causes of inducing DO [148]. Cytokines and oxidative stress from inflammation can affect C-fiber afferents and cause direct sensitization [149]. Histamine released from mast cells can also cause afferent sensitization [150]. Cytokines and oxidative stress during the inflammation also trigger bladder fibrosis and smooth muscle proliferation. Therefore, inflammation could be a cause of OAB and may contribute to bladder hypertrophy in OAB.
In addition, localized immune reactions affect LUTS by sensitizing nociceptors in peripheral afferents and lead to alterations in the CNS. Another route may induce neuro-immune interactions in the CNS using the hypothalamus-pituitary-adrenal axis or the vagal nerve pathway by cytokine released from immune cells, similar to the systemic immune response. The immune system and the CNS may have a bidirectional effect, and LUTD appears to be involved in this process [140, 141]. NGF exerts its action as a peripheral mediator in several inflammatory pain disorders. It is synthesized in the bladder and transmits signals to the CNS through the afferent nerve [151]. In an experimental study, intravesical instillation of exogenous NGF stimulated afferent firing and produced bladder overactivity, and this process could be blocked by anti-NGF treatments [152]. Stretching of the urothelium seems to generate NGF in the bladder. Increased NGF may play an important role in inducing urgency in OAB [153].
Finally, it has been reported that prostatic inflammation is an important factor of LUTS in patients with BPH and that the degree of prostatic inflammation in BPH specimens is associated with the severity of LUTS [154, 155]. Accordingly, animal models of prostatic inflammation induced by chemical irritation in rats or prostatic epithelium-specific deletion of E-cadherin in mice showed that prostatic inflammation-induced bladder overactivity due to prostate-to-bladder pelvic organ cross-sensitization through afferent hyperexcitability, which is mediated at least in part by NGF upregulation in the bladder [156-159]. Thus, local inflammation in the prostate seems to be an important factor inducing male LUTS in BPH patients.
PHARMACOLOGICAL THERAPIES IN OAB AND MECHANISMS OF ACTION
Antimuscarinics
ACh is secreted from the neuromuscular junction of parasympathetic nerves and acts on muscarinic receptors to control the human bladder. Muscarinic receptors are found in the urothelium, interstitial cells, afferent nerves, and detrusor muscles. There are M1–M5 receptor subtypes in the human bladder, and M2 receptors are the most abundant in detrusor muscles, but M3 receptors are predominantly involved in detrusor contraction. Activation of M2 and M3 mediates contraction of detrusor smooth muscle [160-162].
Antimuscarinics are used to treat OAB/DO, inhibiting contraction mediated by M2 and M3 receptors by competitively blocking postjunctional antimuscarinic receptors. This mechanism is appropriate to explain the effect of antimuscarinics on urge incontinence. However, considering that most clinically used doses of antimuscarinics have little or no effect on voiding bladder contractions and act during the storage phase, it is reasonable to assume that it acts primarily through the afferent pathway of the bladder [13, 163]. The mechanisms of antimuscarinics working on the afferent pathway may be explained in the following 3 mechanisms [18]; (1) antimuscarinics may block urothelial ACh generated by mechanical or chemical stimuli in the process of stimulating the afferent nerve indirectly and directly via ATP, (2) ACh from nonneuronal sources can potentiate the afferent noises caused by spontaneous myogenic contractions generated during bladder filling, and antimuscarinics seems to inhibit this process and (3) under neurogenic DO conditions such as nerve injury, spontaneous myogenic contraction may be enhanced by ACh, which is released by neuronal sources as well as nonneuronal sources, and antimuscarinics may suppress the increased afferent activity (Fig. 2).
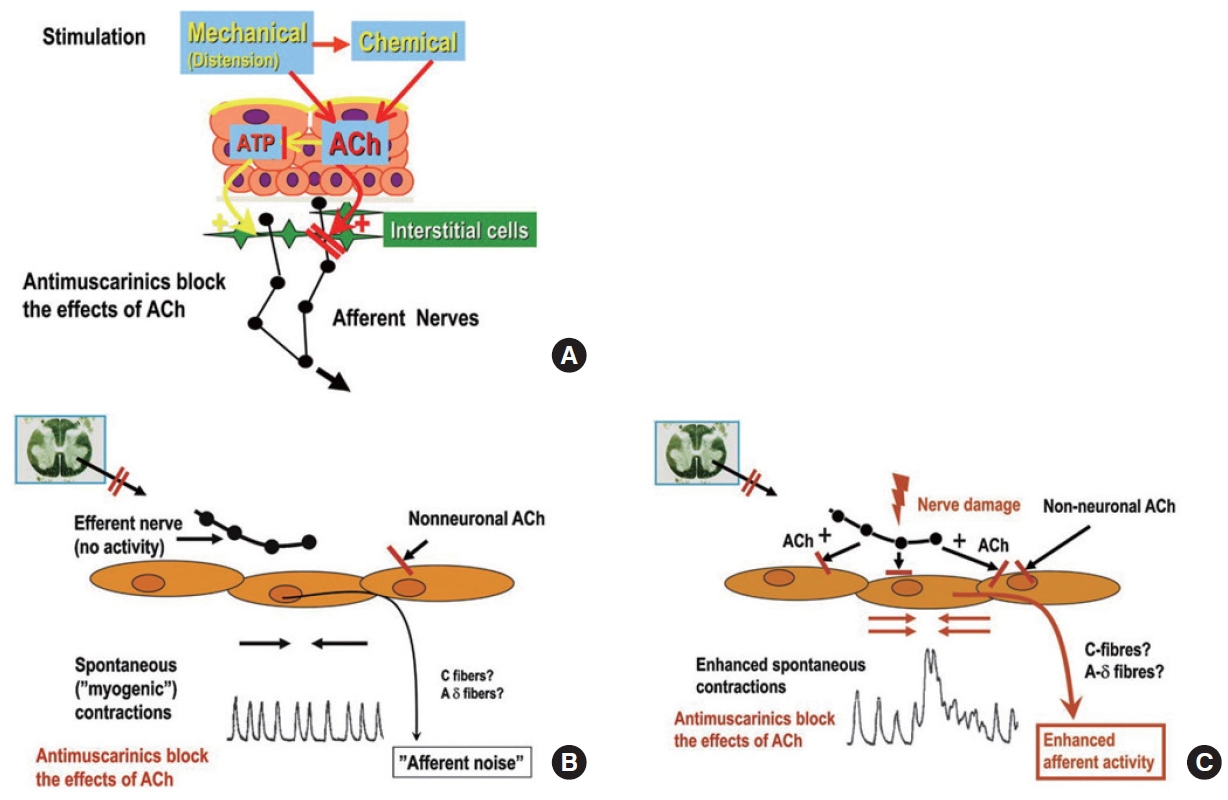
Pathophysiology of autonomous activities (afferent noises) in overactive bladder and antimuscarinic actions on acetylcholine (ACh) effects. (A) Bladder distension (mechanical) or chemical stimulation of the bladder results in urothelial ACh release, which stimulates afferent nerves directly or indirectly via adenosine triphosphate (ATP) release. (B) During bladder filling, spontaneous myogenic contractions, which can be enhanced by non-neuronal ACh release, may happen. Antimuscarinics can block ACh effects. (C) Neuronal and nonneuronal ACh release can increase spontaneous myogenic contractions, which enhance afferent activity. Antimuscarinics can block these enhanced ACh activities. Reprinted from Andersson KE. Eur Urol 2011;59:377-386 [18], with permission of Elsevier.
Given that the concentration of the administered antimuscarinic agent exceeds the therapeutic window, the antimuscarinics may partially attenuate the concentration of released ACh for muscle contraction during the voiding phase, thereby reducing detrusor contraction [18]. These effects may increase residual urine volume or lead to urinary retention. However, at appropriate doses, the combined use of α-blockers and antimuscarinics in male patients with a moderately enlarged prostate (up to 75 g) is safe, even in patients with a post-micturition residual urine volume of up to 150 mL [164].
In idiopathic DO, the evidence is insufficient to draw a certain conclusion about changes in the expression of M2- and M3 muscarinic receptors and changes in functions such as receptor sensitivity [18]. In neurogenic DO, there was a report of decreased numbers of muscarinic receptors and increased receptor sensitivity although the results of a clinical study concluding that a higher concentration of medication was needed for optimal effects in patients with neurogenic DO seem to contradict this finding. However, considering the treatment goal of neurogenic DO is to reduce the intravesical pressure to prevent upper urinary tract damage, it seems inappropriate to compare the concentrations of the OAB treatment regimen under non-equivalent conditions [165, 166].
Reduced detrusor responses by intramural nerve stimulation and postjunctional supersensitivity to ACh have been demonstrated in the BOO experiment using pigs and patients with BOO and DO. In the partial BOO model using rats, the muscarinic receptor-coupled RhoA/Rho-kinase pathway was activated as a mechanism to compensate for bladder emptying [167, 168]. M3 receptors predominantly mediate detrusor contractions in BOO-induced hypertrophied rat bladder, and another study also reported that M2 density increased while M3 density decreased [169, 170]. However, the relevance of these findings associated with OAB pathophysiology has not yet been elucidated.
Finally, muscarinic receptors are abundant in the CNS and involved in memory and cognitive functions. Most antimuscarinic drugs in clinical use can pass through the BBB whereas solifenacin, trospium, and darifenacin have been shown to have little or no risk for cognitive decline compared to oxybutynin in healthy older adults with OAB. However, the role of β3-AR agonists in OAB treatment has been highlighted as various studies have revealed the risk of exacerbations in cognitive function in elderly patients with Alzheimer’s disease due to anticholinergic accumulation [171].
Phosphodiesterase Inhibitors
In the lower urinary tract, NO is involved in several key functions. In the bladder, NO is synthesized in the urothelium, detrusor muscle, and nerves and can regulate detrusor smooth muscle tone, bladder compliance, and micturition reflex. In the urethra and the prostate, NO is generated by non-cholinergic parasympathetic nerves, vascular endothelial cells, and smooth muscle cells and is implicated in the control of urethral tone, continence mechanisms, and regulation of the secretory function of the prostatic gland. NO diffused into the peripheral tissue activates guanylyl cyclase, which catalyzes guanosine triphosphate into cyclic guanosine monophosphate (cGMP), and the increased cGMP activates protein kinase G, leading to smooth muscle relaxation [172]. In this process, phosphodiesterase inhibitors (PDE5i) inhibit the degradation of cGMP and promote the downstream signaling process from NO. There has been some evidence that PDE5i improve urinary tract symptoms in men with erectile dysfunction and LUTS [173-175]. In the BOO model using rats, bladder muscle relaxation was facilitated by increased NO signaling [176]. Adenylyl cyclase is an enzyme that catalyzes the metabolism of ATP to cyclic adenosine monophosphate (cAMP), and increased cAMP relaxes the detrusor muscle strip of pigs [177]. PDE4 inhibitors suppress cAMP metabolism, thereby preventing the activity of myosin light-chain kinase via an increase in protein kinase A to induce smooth muscle relaxation [178]. In the studies using BOO rat model, selective PDE4 inhibitors effectively suppressed DO without effects on bladder contractility [179, 180]. However, as a clinical regimen, PDE4 inhibitors have the problems of most implicated gastrointestinal side effects such as nausea and vomiting.
Botulinum Neurotoxin A
Botulinum Neurotoxin A (BoNT-A) has been used to treat a variety of LUTD conditions since it was first used in 1988 to treat detrusor sphincter dyssynergia in men with SCI. It shows successful results not only in the treatment of neurogenic DO caused by SCI, but also in patients with idiopathic OAB and DO.
BoNT-A inhibits the release of ACh and other neurotransmitters from efferent nerve terminals including pre- and postganglionic parasympathetic nerve terminals [181]. In the bladder, it inhibits the release of ACh from the efferent nerve, reducing detrusor contractility during micturition, inhibiting vesicular noradrenaline release, preventing α- and β3-AR activation, and additionally affecting bladder neck contracture and detrusor relaxation [182]. BoNT-A may also act through the afferent nerve pathway. In an investigation of the patients with neurogenic DO injected by BoNT-A into the bladder, a significant decrease in M2-, M3 muscarinic receptors as well as P2X2- and P2X3 purinergic receptors in the muscle layer was observed [183]. Considering that the bladder of P2X3-null mice showed significantly decreased sensitivity to bladder filling, ATP and its receptor, P2X3, may contribute to the pathogenesis of OAB by playing an important role in the modulation of the urinary bladder volume reflex [184]. A biopsy conducted after BoNT-A injection into the bladder of neurogenic and idiopathic DO patients demonstrated decreased expression of P2X3 and TRPV1 immunoreactivity in afferent nerve fibers. Additionally, the degree of decrease in P2X3 and TRPV1 was correlated with improvement in frequency and urgency [185]. ATP and neurotrophins released from the urothelium were suppressed, and NO secretion was increased after BoNT-A treatment. Based on these results, BoNT-A seems to inhibit the release of ATP and neurotransmitters related to afferent sensitization in the afferent nerve and urothelium [182].
β3-AR AGONIST ACTION ON OAB AND ITS SITE OF ACTION IN VARIOUS PATHOLOGIC CONDITIONS
β-ARs have 3 subtypes (β1, β2, and β3) in both the detrusor muscle and the urothelium [186]. In an immunohistochemistry study, β3-ARs were found not only in the detrusor smooth muscle in rat and human bladder but also in the urothelium [187], interstitial cells in the suburothelial layer, afferent nerve [187, 188], and recently in L6-S1 dorsal root ganglion (DRG) neurons [182]. Immunoreactivity of β3-ARs was also found in small-diameter neurons in the major pelvic ganglion of rats [189]. These results suggest that β3-ARs are involved in a neural circuit that controls afferent outflow and sensation. An activation of β3-ARs catalyzes the conversion of ATP to cAMP through adenylyl cyclase activation, which decreases intracellular Ca2+ concentration and results in the relaxation of detrusor smooth muscle [162]. In addition to the cAMP-dependent pathway, potassium channels may also be involved in β3-AR agonist-induced detrusor relaxation [190].
Activation through β3-AR agonists increased bladder capacity without changing voiding pressure and residual urine volume during the voiding phase [191-193]. Also, under isovolumetric conditions, the frequency of rhythmic bladder contraction was reduced without suppressing contraction amplitude [194]. These results suggest that the mechanisms of β3-AR agonists may be involved in bladder relaxation during the storage phase without affecting the voiding phase [195, 196].
β3-AR agonists inhibited spontaneous myogenic contractions and non-voiding contractions, which enhance afferent activities [197]. Bladder distention initiated a low threshold mechanoreceptive Aδ-afferents, and mirabegron, a β3-AR agonist, reportedly inhibited mechanosensitive (Aδ) afferents activity, possibly associated with inhibiting bladder microcontractions. In another study, CL316,243, a β3-AR agonist, inhibited filling-induced activity, which may involve Aδ- and C-fiber afferents, but predominantly Aδ-fibers [198]. The β3-AR agonist is also involved in inhibiting autonomous bladder contraction, but is thought to have little effect on coordinated voiding contraction induced by ACh or ATP. These results could explain why the β3-AR agonist has little effect on bladder emptying [199]. Tolterodine, an antimuscarinic, reduced the amplitude and frequency of nonvoiding contractions (NVCs), whereas mirabegron mainly affected frequency and tolterodine reduced voiding contraction in a dose-dependent manner. β3-ARs at parasympathetic terminals directly inhibited the cholinergic pathway related to excitatory motor drive [200]. β3-AR agonists also inhibited cholinergic transmission by adenosine-induced retrograde activation of prejunctional A1 receptor, where equilibration nucleoside transporters 1 (ENT1) was involved in adenosine release from detrusor smooth muscle. In addition, β3-AR agonists exerted a fine control of the sensory bladder drive, which occurred during the storage phase by adenosine released from urothelium via ENT1. Thus, β3-AR agonists, which may inhibit endogenous adenosine-mediated cholinergic neurotransmission, increase bladder capacity during the storage phase and increase micturition interval without affecting micturition pressure or residual urine volume [201].
Bladder Outlet Obstruction
The expressions of the β3-ARs subtype in the human detrusor muscle were not different regardless of BOO [202]. In the BOO model, mirabegron decreased the frequency of NVCs as well as spontaneous contractile activities [203]. Previous studies have found spontaneous contractile activity in the mucosa of guinea pigs and pig bladders, and one possible source of this activity is suburothelial interstitial cells [204-206]. These spontaneous contractile activities from interstitial cells partially contributed to enhanced bladder afferent interactions [197]. Therefore, one of the possible mechanisms of β3-AR agonist action may be the inhibition of spontaneous contractile activities through β3-ARs of interstitial cells in the bladder. Mirabegron inhibits afferent activities, specifically Aδ-fiber afferents, enhanced by myogenic microcontraction in both normal and BOO bladders in rats. This mirabegron-mediated inhibition is considered to occur in β3-AR located in afferent nerves [198]. A recent study showed that vibegron could partially inhibit mechanosensitive afferent transduction through Aδ- and C-fibers by reducing myogenic contractile activities in the BOO-induced hypertrophied bladder in rats [207].
Bladder Ischemia
In a bladder ischemia model using rats, the relaxation response of the isolated detrusor strip to isoprenaline, a non-selective β-AR agonist, and salbutamol, a β2-AR agonist, did not change, whereas the relaxation response to BRL 37,344, a selective β3-AR agonist, was increased [208]. In another study for chronic bladder ischemia, long-term treatment with mirabegron prevented bladder hypertrophy and fibrosis. These results suggest that β3-ARs may be a potential therapeutic target in chronic ischemia-related bladder dysfunction [209].
Neurogenic LUTD
In normal bladder, β3-AR agonist exerts its action by inhibiting ACh release in parasympathetic nerves and suppressing the afferent nerve pathway from the urothelium to the afferent nerves [210]. Aδ-fiber afferents mainly control normal micturition. However, after SCI, micturition depends on DO, resulting from increased excitability of capsaicin-sensitive C-fiber afferents [211], where neurotrophic factors such as NGF and BDNF (brain-derived neurotrophic factor) are involved [212]. In SCI mice, pretreatment with capsaicin suppressed NVC during filling but did not affect decreased bladder capacity, compliance, high voiding pressure, and poor voiding efficiency. These results show that DO and micturition reflex are triggered by different afferent mechanisms [213]. When vibegron was administered to SCI mice, NVCs were suppressed, and the interval until NVCs occurred was prolonged, but there was no change in other cystometric parameters. These results indicate that vibegron, a β3-AR agonist, inhibits capsaicin-sensitive C-fiber afferents in SCI mice [214]. In contrast, mirabegron administered to SI rats suppressed mechanosensitive afferent activity related to rhythmic bladder contraction, in which the inhibition of Aδ-fiber afferents acted predominantly rather than C-fiber afferents [29].
Overactive Bladder
Mirabegron reduced carbachol-induced detrusor muscle tone equally in 3 experimental groups of normal patients, patients with BOO only, and patients with BOO-induced DO in a concentration-dependent manner [215]. In addition, β3-AR agonists suppressed detrusor contraction induced by endogenous ACh by 67%, but did not suppress detrusor contraction induced by exogenous ACh administration (only 25% reduction). These results indicate may suggest that the β3-AR is present in ACh-containing nerves, especially parasympathetic nerves, and is involved in detrusor relaxation mediated by prejunctional mechanisms. [188, 200, 216]. Considering that the β3-AR agonists did not decrease the micturition pressure during the voiding phase, the β3-AR-induced therapeutic mechanism may be explained by the suppression of the pathologically increased cholinergic tone during the filling phase in OAB.
When vibegron, a new β3-AR agonist, was administered to the OAB rat model, increased expression of urothelial β3-AR, increased bladder capacity, and decreased threshold pressure were confirmed without affecting contractile function. Based on these results, vibegron may work through 2 pathways; (1) a mechanism that inhibits ACh release from the cholinergic efferent nerve in the detrusor and (2) an afferent inhibition mechanism through urothelial β3-AR [217] (Fig. 3).
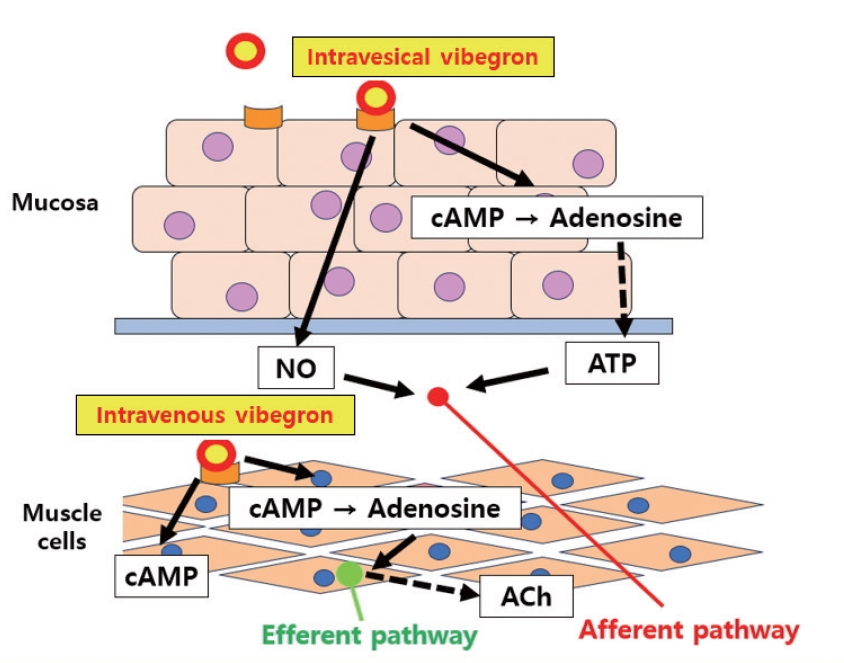
Putative mechanisms of intravesical and intravenous vibegron on bladder function. Vibegron may work through 2 pathways; (1) a mechanism which inhibits ACh released from the cholinergic efferent nerve in the detrusor and (2) an afferent inhibition mechanism through urothelial β3-adrenergic receptors. cAMP, cyclic adenosine monophosphate; ATP, adenosine triphosphate; Ach, acetylcholine.
Effects on CNS
In a chemically induced DO mouse model using KCl, repetitive bladder insults induced an increase in NVCs and decreases in intercontraction intervals, bladder capacity and voiding efficiency without changes in micturition pressure in CMG. mRNA expressions of β3-ARs, M2-, and M3 muscarinic receptors, and P2X purinergic receptors were upregulated in the urothelium of the bladder, which indicates hyperexcitability of bladder afferents. In the L6 spinal cord, immunoreactivities of CX3C motif chemokine receptor 1 (CX3CR1), glial fibrillary acidic protein (GFAP), and C-C chemokine receptor type 2 (CCR2), which are implicated in the neuroinflammation (neuro-glial interactions), were elevated. These results suggest that chronic noxious stimuli of bladder afferents may induce neural remodeling of CNS; in other words, central sensitization. Long-term continuous treatment of mirabegron reduced NVCs and improved bladder capacity and voiding efficiency. In addition, immunoreactivities of CX3CR1, GFAP, and CCR2 were significantly decreased by continuous administration of mirabegron compared to the sham or treatment cessation group, which indicates that continuous treatment may prevent central sensitization [9]. This central sensitization mechanism was probably possible because mirabegron acts on β3-ARs in the urothelium and preganglionic cholinergic nerves, thereby continuously suppressing afferent signals and cholinergic efferent transmission, which could influence the CNS activation mechanisms.
There is still insufficient evidence of whether β3-ARs participate in neural remodeling by working directly on DRG or CNS. There were some studies on the existence of β3-ARs on DRG [182], and CNS [218, 219]. Although a detailed role has not been revealed yet, β3-ARs may be involved in controlling depression in the frontal cortex and hippocampus [218-220], and interact with serotonin receptors such as 5-HT1A, 5-HT2A, and 5-HT3 [221, 222]. β3-ARs presented in the locus coeruleus are also associated with norepinephrine secretion [223]. There was a study showing a correlation of β3-ARs with neuroinflammation mediated by ATP in DRG [224], but a recent study revealed that only peripheral β3-ARs are involved in these mechanisms of neuroinflammation [225].
CONCLUSIONS
Several hypotheses have been suggested for the mechanisms involved in OAB development, including those of neurogenic, myogenic, and urothelial origin; however, it is difficult to explain them using just one hypothesis as it seems that they are tangled and influence each other. Because the pathologic etiologies that cause OAB are diverse, the OAB phenotype is also assumed to be multifactorial, although no phenotype standardization exists. In fact, the available treatment regimens are still not sufficient. Each drug has different mechanisms of action, and the drug effects may work differently depending on the underlying pathologies, although the same drug is used. If continued clinical and basic research increases our understandings of the action mechanisms in OAB medications including β3-AR agonists, better treatment approaches can be achieved to implement the pathogenesis-oriented therapy for OAB patients who do not respond to or have little effects by conventional treatments.
Notes
Grant/Fund Support
The research work by authors has been supported by grants from the National Institutes of Health (R01 DK129194 to NY and SK; R01 DK133434 to AMR and NY; R01 DK134580 to ZW & NY).
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION STATEMENT
·Conceptualization: JK, SK, AMR, PT, NY
·Data curation: JK, KJC, MH, KM, TK, NY
·Formal analysis: JK, KJC, MH, NY
·Funding acquisition: NY
·Methodology: JK, DYK, KJC, MH, ZW, NY
·Project administration: JK, DYK, SK, AMR, PT, NY
·Visualization: JK, KJC
·Writing-original draft: JK
·Writing-review & editing: JK, DYK, NY