Oxidative Stress and Its Relation to Lower Urinary Tract Symptoms
Article information

Abstract
The aim of this review is to discuss how to link lower urinary tract symptoms (LUTS) and oxidative stress (OS) and to define relevant targets for therapeutic intervention. Narrative review based on published literature. Many of the multifactorial pathophysiological mechanisms behind LUTS can initiate reactive oxygen species (ROS) generation. Assuming that OS is a consequence rather than a primary cause of LUTS it seems reasonable to identify both the disease mechanism initiating LUTS, and the source of ROS involved. There are many possible sources of ROS overproduction, but the NADPH oxidase (NOX) family of enzymes is the primary source; NOX activation in turn, may result in the activation of secondary ROS sources, i.e., ROS-dependent ROS production. Selective NOX inhibition therefore seems an attractive therapeutic strategy in LUTS treatment. The finding of NOX2 localization to centers in the brain associated with micturition control, opens up for further studies of NOX involvement in the central control of micturition, normally and in disease. Further information on the localization of the different isoforms of NOX in the LUT e.g., the bladder wall and its components and the prostate, is desirable. To optimize treatment, the pathophysiological mechanism initiating LUTS, and the activated isoform of NOX, should be identified. Unfortunately, in most cases of LUTS this is currently not possible. Even if selective NOX inhibitors have entered the clinical trial stage for treatment of disorders other than LUT dysfunction, their efficacy for LUTS treatment has to be demonstrated. If this can be achieved, an attractive approach would be combination of selective NOX inhibition with established drug therapies.
INTRODUCTION
Lower urinary tract symptoms (LUTS) have been associated with a large number of diseases [1], and so has oxidative stress (OS). As pointed out by Wu et al. [2], aging and major chronic diseases are major risk factors for LUTS. They suggested that OS might be a candidate mechanism linking LUTS to these entities and this could make OS a therapeutic target for LUTS. Both aging and chronic diseases involve multiple pathophysiologic mechanisms (Fig. 1). It is obvious that some of them with or without involvement of OS can cause LUTS (Fig. 2). A number of questions has to be answered before any realistic therapeutic approaches can be designed. Are the mechanisms initiating LUTS also initiating OS? Is OS a consequence, not a cause of LUTS, or is OS initiating LUTS (Fig. 2).
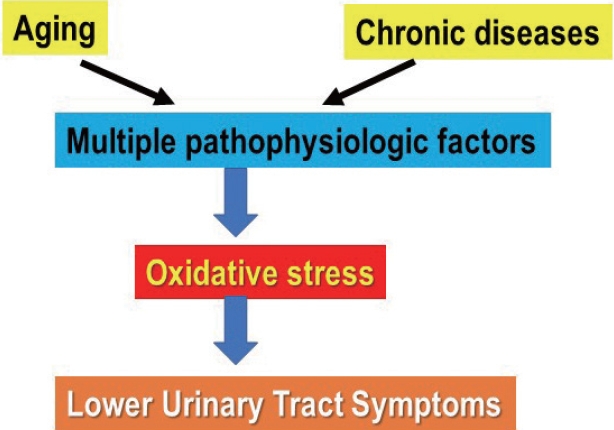
Aging and chronic diseases may involve many pathophysiologic factors that can produce oxidative stress

Multiple pathophysiologic factors may initiate lower urinary tract symptoms (LUTS) with or without simultaneously activating oxidative stress (OS); they may activate OS before LUTS or LUTS before OS. LUTS and OS may interact, OS enhancing LUTS or LUTS enhancing OS.
A causal molecular mechanism for most common diseases is not known and most diseases, including e.g., OAB, are multifactorial [3]. It is often convenient to look at OS as a final common destructive pathway involved in aging processes and tissue destruction and degeneration in chronic diseases. However, it should be remembered that reactive oxygen species (ROS) causing OS, “at every concentration, low or high, can serve many essential signaling and metabolic functions” [4]. Is ROS then a reasonable target for drugs aimed for treatment of LUT disorders? If so, which are the reasons why antioxidants have not been demonstrated to ameliorate diseases where ROS is implicated [4-7]?
Recent developments in drug research aiming at finding drugs that are effective to treat OS have focused on identifying and selectively modulating ROS enzymatic sources that produce the ROS having the harmful effects [4]. But how are the ROS enzymatic sources activated? Ghezzi et al. [5] stated: “Today it is a challenge to find a disease for which a role of OS has not been postulated.” It seems easier to identify factors/mechanisms that are associated with LUTS than those activating NOX. Assuming that a LUTS associated mechanism is the stimulus that causes the generation of ROS and the imbalance between oxidants and antioxidants (Fig. 2), it seems reasonable to try to define not only this mechanism, but also which ROS species involved.
The aim of this review is to discuss possible links between LUTS and OS and to define relevant targets for therapeutic intervention.
LOWER URINARY TRACT SYMPTOMS
Generation of LUTS
LUTS can be divided into 3 categories [8]: storage (irritative) symptoms, including urgency, frequency, nocturia, and urgency incontinence, i.e., the overactive bladder–OAB-syndrome; voiding (obstructive) symptoms, comprising reduced force of stream, hesitancy, inability to empty the bladder, and straining; and postmicturition symptoms with feeling of incomplete emptying and postmicturition dribble. Most of these symptoms have been suggested to be age-dependent and attributed to various factors, including reduced bladder capacity, changes in bladder sensation, detrusor overactivity (DO; on urodynamic investigation) and OAB, and also to involve OS.
As mentioned, the pathophysiology of OAB and other LUTS is multifactorial [3] and most often seems to involve disturbances of afferent signaling from the LUT. Afferent signaling may be regarded as a final common pathway for LUTS and its origin has been extensively discussed [9-11]. It has also been suggested as a common treatment target for drugs aiming at LUT disorders [12]. Basically, 2 main afferent signaling pathways can been defined: the mucosal and the myogenic [9]. In the myogenic pathway the intrinsic rhythmicity of detrusor muscle cells activates afferent nerves, and in the urothelial pathway the urothelium (by release of mediators) activates afferent nerves in the lamina propria. Both pathways can generate afferent activity in both Ad-fibers involved in normal bladder contraction and DO, and in Cfibers mediating sensory perceptions–urge and urgency, and both can interact and be modulated by structures in the lamina propria (e,g., interstitial cells). In the lamina propria, afferent nerves form a plexus that lies immediately beneath the epithelial lining. This plexus is particularly dense in the bladder neck and the trigone [13,14] and is important for urothelial signaling [15]. The nerves release numerous ‘sensory” peptides (e.g., substance P, calcitonin gene-related peptide, vasoactive intestinal polypeptide, enkephalins, and cholecystokinin), and contains multiple receptors (e.g., transient receptor potential channels, purinoceptors, tachykinin, and prostanoid receptors) which may be therapeutic targets. It is most likely that a cascade of inhibitory and stimulatory transmitters/mediators are involved in the transduction mechanisms underlying the activation of afferent fibers during bladder filling [9]. Even if each of these signal mediators can be a target for drugs meant for treatment of LUT disorders, it is reasonable to assume that more than one is contributing, which means that drugs selectively blocking only one mechanism will have a limited effect. It has been suggested that in e.g., the urothelium there is an aging-related increase in OS with excessive generation ROS which by damaging multiple components of the LUT system can contribute to LUTS [16]. However, there may be other structures in the bladder wall involved in LUTS generation with consequent production of ROS. Is any of these structures more important than others as ROS generator and can it be targeted selectively?
OXIDATIVE STRESS
Oxidants
OS has been defined as “an imbalance between oxidants and antioxidants in favor of the oxidants, leading to a disruption of redox signaling and control and/or molecular damage” [17,18]. Oxidants contributing to the stress include ROS, reactive nitrogen species (RNS) and free radicals, such as superoxide anion radical (O2 •−) and hydroxyl radicals (•OH), but also nonradicals, such as H2O2, nitric oxide (NO), peroxynitrite, and hypochlorous acid. The only enzyme family known to produce ROS as its sole function are NADPH oxidases (NOX). However, many other enzymes, e.g., xanthine oxidase (XO), monoamine oxidases, lysyl oxidases, lipoxygenase (LOX), and cyclooxygenase, produce superoxide and H2O2 secondary to their primary metabolic function [19].
ROS is often used as a generic term including all oxidants, but all ROS molecules are not the same. It has been underlined that each of these molecules is a distinct chemical entity with its own reaction preferences, kinetics, rates, site of production and degradation, and diffusion characteristics in biological systems [20]. The implications of this are that the biological impact of ROS will be critically dependent on the particular molecule(s) involved, and on the microenvironment and physiological or pathological context in which it (they) is (are) being generated [20].
Antioxidants
The antioxidant defense systems have been developed to eliminate ROS and include enzymatic and nonenzymatic pathways. The primary enzymes are superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GSH-Px) [2]. SODs are the only enzymes that interact with superoxide specifically and control the levels of ROS and RNS. They also serve as key regulators of signaling [21]. SODs convert superoxide to H2O2, which is decomposed to water and oxygen by CAT. SODs prevent the accumulation of superoxide to damaged tissue and inactivate proteins containing iron–sulfur clusters. SOD1 is mainly located in the cytosol and mitochondrial intermembrane space; SOD2 is located in the mitochondrial matrix while SOD3 is located in the plasma membrane. SOD and CAT activities are associated with the shift from the compensated to decompensated function of the bladder [22]. GSH-Px converts peroxides and hydroxyl radicals into nontoxic forms by the oxidation of reduced glutathione (GSH) into GSH disulfide and then reduced to GSH by GSH reductase. Other antioxidant enzymes are GSH-S-transferase and glucose-6-phosphate dehydrogenase. The nonenzymatic antioxidants bilirubin, α-tocopherol (vitamin E), and β-carotene are molecules that interact with ROS and RNS and terminate the free radical chain reactions They are present in blood while albumin and uric acid account for 85% of antioxidant capacity in plasma [23].
SOURCES OF ROS
NADPH Oxidases
ROS can be produced by several sources, including mitochondrial ROS formation, XO, uncoupled NO synthase and NOX [24]. Activation of ROS generating enzymes can result in the activation of secondary ROS sources, i.e., ROS-dependent ROS production [25]. One of the most important sources of ROS is the family of NOX [4,26]. These enzymes are widely distributed in the body including various types of smooth muscle and epithelium [26]. NOX mediates the transfer of electrons from cytosolic NADPH, through flavin adenine dinucleotide to penetrate the membrane, via hemes, to oxygen, leading to superoxide generation in the cytoplasm. The NOX family is known to have several subtypes: NOX 1-5; and Duox 1 and 2, with different tissue and cellular distributions [4,27-30]. These enzymes have multiple important physiological functions in the cell and therefore NOX inhibition may perturb normal physiology in unpredictable ways. Is then inhibition of NOX subtypes a possible way to achieve effects that can be beneficial in LUT disorders without negatively affect other organ functions?
Mitochondria
Mitochondria are an important source of ROS within most mammalian cells and superoxide (O2 •−) is the proximal mitochondrial ROS [20,30,31]. Mitochondrial ROS production is important in redox signaling from the organelle to the rest of the cell but can also contribute to mitochondrial damage in a range of pathologies. Mitochondria are considered to generate 95% of all cellular energy and are the major consumers of cellular oxygen. Therefore, it is not surprising that they are significantly impacted by hypoxia and ischemia. Reduced levels of oxygen result in augmented ROS production, decreases energy production and changes in mitochondrial morphology.
Soler et al. [32] studied a mutant mouse model with a deficiency of the Immp2l protein and showing abnormal signal peptide sequence processing of the mitochondrial proteins, cytochrome c1 and glycerol-3-phosphate dehydrogenase. Bladder mitochondria in these mutant mice manifest hyperpolarization, increased superoxide ion generation and increased adenosine triphosphate, probably due to a high mitochondrial respiration rate. The mutant mice had disturbances of bladder function including pronounced voiding difficulty and straining when initiating micturition. They had higher postvoid residual volumes than control, but in vitro the detrusor contractile responses to carbachol and electrical field stimulation were similar in mutant and wild type mice. Mitochondrial ROS production may seem an attractive target for treatment. However, currently it is not considered realistic/safe to target mitochondrial ROS pharmacologically in humans [4].
Sources of NOX Relevant for Micturition.
NOX 2 has been localized in in the periaqueductal gray (PAG) and Barrington’s nucleus (pontine micturition center, PMC), important structures in the micturition reflex [33]. Wu et al. [33] showed that in these parts of the brain NOX 2 protein produces a significant amount of ROS suggesting that NOX-associated ROS production is important in the central nervous bladder control. Superoxide generation in PAG and PMC was significantly suppressed by the NOX inhibitor diphenyleneiodonium (DPI) and also reduced by the NOX-2 specific inhibitor GSK2795039.
Information on the localization of NOX isoforms in the LUT is limited. NOX 1 and NOX 5 are found in the prostate [34] but the NOX isoforms located to the bladder wall and its components do not seem to have been specifically studied. Information on NOX expression in LUT dysfunction is generally lacking, but NADPH-dependent superoxide production has been reported in bladder urothelium and may lead to oxidative damage and contribute to bladder overactivity [16].
SELECTIVE NOX INHIBITORS
The nonselective ROS scavenging effects of antioxidants interfere with both physiological and pathological ROS. It is therefore necessary that the disease relevant enzymatic sources of ROS are identified and selectively targeted, leaving physiological ROS signaling through other sources intact [35]. NOX (NOX1-5 and Duox1-2) produce ROS as their sole function, whereas most other enzymatic sources only produce ROS as a by-product or upon biochemical uncoupling or damage. This qualifies NOXs as the main potential drug-target candidates in diseases associated with dysfunction in ROS signaling. As a reflection of this, several more or less selective NOX inhibitors are in development [36]. Recently, the World Health Organization approved a new stem, “naxib,” which refers to NADPH oxidase inhibitors, and thereby recognized NOX inhibitors as a new therapeutic class [35].
According to Dao et al. [24] NOX-specific and isoform-selective pharmacological NOX inhibition using small molecules is in principle achievable since these compounds show different apparent NOX isoform selectivity profiles. However, only a few compounds are claimed to be specific NOX inhibitors in preclinical studies, including GSK2795039 which selectively inhibits NOX2 [37], GLX7013114 for NOX4 [38], and Ewha-18278 (APX-115) for NOX1, NOX2, and NOX4 [39]. Clinical trials with setanaxib (initially known as GKT137831) inhibiting NOX1 and NOX4, have been performed [35], demonstrating excellent tolerability and reduction of various markers of chronic inflammation.
BLADDER ISCHEMIA, LUTS AND ROS–THERAPEUTIC APPROACHES
Bladder ischemia is a common cause of LUTS, especially in the elderly and in patients with bladder outlet obstruction [40-42]. Clinically, treatment has so far been focused mainly on improving bladder perfusion [42,43]. Even if the α1-adrenoceptor blocker, silodosin, the phosphodiesterase type 5 inhibitor, tadalafil, the β3-adrenoceptor agonist, mirabegron, and the free radical scavenger, melatonin, were unable to prevent the development of neointimal hyperplasia and consequent luminal occlusion in animal models [43], they all exerted a protecting effect on urodynamic parameters, and on some of the functional and morphological changes of the bladder demonstrable in vitro.
Generation and release of ROS associated with ischemia and hypoxia/reoxygenation is known as one of the most deleterious causes of oxidative damage with changes in the structural integrity of smooth muscle cells, cellular organelles, microvasculature, and nerve fibers, leading to dysfunction [44-46]. Three potential sources of ROS have been proposed to be responsible for this release: mitochondrial complex I, XO, and NOX [25]. Free radical scavengers, such as melatonin and coenzyme Q-10, have been shown to prevent free radical damage associated with hypoxia-reperfusion injury and chronic ischemia in the rat bladder [46], but so far therapy directed at the different isoforms of NOX have apparently not been tested.
Preventing ROS Damage
Identifying the pathophysiological mechanism and intervening pharmacologically seems to be the logical and appropriate way of treating lower urinary tract (LUT). Considering the multifactorial pathophophysiology of LUTS with a large number of possible disease mechanisms, this may be possible only in some cases. As mentioned, treatment of LUTS caused by bladder ischemia can be directed at relieving atherosclerotic obstruction of the arterial blood flow or inducing vasodilation to improve blood perfusion to the organ [40-43]. Since bladder ischemia may also be the initiator of increased ROS production and OS, using antioxidants and scavengers of free radicals to protect against oxidative damage should be reasonable additional approaches provided that the ROS species involved can be identified and selective organ effect obtained. NOX-specific and isoform-selective pharmacological NOX inhibition using small molecules is in principle achievable, but only a few compounds are claimed to be specific NOX inhibitors in preclinical studies. The different isoforms of NOX are widely distributed in the body, but it seems that the isoforms in the LUT, specifically the bladder, have so far not been defined [35]. This is information urgently required if LUTS treatments based on defined disease mechanisms (e.g., ischemia) or positive responses to established drugs can be combined with effective NOX inhibition.
Repairing ROS Damage
The role of NO as a relaxing factor in bladder, bladder neck, urethra, and prostate smooth muscle is well established. The target for NO is its intracellular receptor, soluble guanylate cyclase (sGC), which on stimulation produces cyclic GMP (cGMP). The relaxing effect of cGMP is terminated by cGMP-specific 3ʹ,5ʹ-cyclic phosphodiesterase (PDE5), and to some extent, by PDE6 and PDE9A. The clinical efficacy of PDE5 inhibitors (PDE5Is) to ameliorate LUTS associated with BOO is well documented and their effects include alleviation of OS [47]. The effects of the PDI5 inhibitors are dependent on NO production. If NOS activity is reduced, can there be other ways to stimulate/activate cGMP and thereby restore and repair ROS-induced damage? Attempts have been made to find drugs that act directly on sGC in an NO independent manner [48]. YC 1 was the first NO independent and haem-dependent sGC stimulator but owing to its poor adverse effect profile, novel compounds with increased potency and selectivity were developed [48-51] primarily for treatment of cardiovascular diseases: NO independent, haem-dependent stimulators of sGC (BAY 41–2272, BAY 41–8523, BAY 63–2521, and BAY 60–4552) and NO independent, haem-independent sGC activators (HMR 1766, BAY 58–2667, and BAY 60–2770). These drugs may be effective for patients who are refractory to PDE5Is.
One of the sGC activators, cinaciguat (BAY 58-2667), was recently studied by Zabbarova et al. [52] who demonstrated that in aged mice/rats, exhibiting histologic, morphologic, and urodynamic features of human BOO associated LUTS, the drug ameliorated these symptoms. Cinaciguat also improved bladder function in cyclophosphamide-induced cystitis in mice [53]. Cinaciguat has been tested in healthy volunteers [54] and in patients with acute decompensated heart failure and was well tolerated [55]. These findings make this drug an interesting candidate for further testing in patients with LUT dysfunction.
CONCLUSIONS
It is reasonable to believe that many of the multifactorial pathophysiological mechanisms behind LUTS can initiate ROS generation, making OS a consequence rather than a primary cause of LUTS. There are many possible sources of ROS overproduction, but NOX enzymes are the primary sources of ROS, and their activation in turn, may result in the activation of secondary ROS sources, i.e., ROS-dependent ROS production. Selective NOX inhibition seems an attractive therapeutic strategy in LUTS treatment, and the finding of NOX2 localization to PAG and PMC opens up for further studies of NOX involvement in the central control of micturition, normally and in disease. Further information on the localization of the different isoforms of NOX in the LUT e.g., the bladder wall and its components and the prostate, is desirable. Since none of the ROS sources act on their own, and different ROS forming enzymes will affect different targets, combinations of different NOX inhibitors could most probably lead to better efficacy and reduced side effects. The pathophysiological mechanism initiating the activation should also be identified and treated. In most cases of LUTS this is not possible, which reflects that established treatment principles often have only modest efficacy. Even if selective NOX inhibitors have entered the clinical trial stage (for disorders unrelated to the LUT) their efficacy for LUT disorder has to be demonstrated. If this can be achieved, the combination of established treatment with selective NOX inhibition seems attractive since an increased efficacy level, compared to what can be obtained with available drugs, might be expected.
Notes
Conflict of Interest
No potential conflict of interest relevant to this article was reported.