Exercise and Neuroinflammation in Health and Disease
Article information

Abstract
Neuroinflammation is a central pathological feature of several acute and chronic brain diseases, including Alzheimer disease (AD), Parkinson disease (PD), amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS). It induces microglia activation, mitochondrial dysfunction, the production of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), pro-inflammatory cytokines, and reactive oxygen species. Exercise, which plays an important role in maintaining and improving brain health, might be a highly effective intervention for preventing neuroinflammation-related diseases. Thus, since exercise can improve the neuroimmune response, we hypothesized that exercise would attenuate neuroinflammation-related diseases. In this review, we will highlight (1) the biological mechanisms that underlie AD, PD, ALS, and MS, including the neuroinflammation pathways associated with microglia activation, NF-κB, pro-inflammatory cytokines, mitochondrial dysfunction, and reactive oxygen species, and (2) the role of exercise in neuroinflammation-related neurodegenerative diseases.
• HIGHLIGHTS
- Neuroinflammation is a central pathological feature of brain disease.
- Neuroinflammation induces microglia activation, mitochondrial dysfunction, the production of NF-κB, pro-inflammatory cytokines, and reactive oxygen species.
- Exercise plays an important role in preventing neuroinflammation.
INTRODUCTION
Inflammation plays a pivotal role during the biological response to defend and support the body following noxious stimuli and conditions such as injury, trauma, and infection [1]. It removes invading pathogens and induces angiogenesis and wound healing [2] through phagocytosis and the activation of inflammasomes, which induce programmed cell death [3] to ultimately facilitate tissue regeneration [4]. However, even though inflammation is beneficial and protective, excessive inflammation can induce tissue damage and lead to the development of pathological diseases [4]. In the brain, the inflammatory response may be beneficial and vital in some circumstances, but can also be harmful by causing acute and chronic brain disorders [1]. Therefore, knowing when inflammation is protective or detrimental could be paramount in understanding brain-related diseases.
Neuroinflammation is an immune reaction that occurs in response to various signals, such as infection, traumatic brain injury [5], autoimmunity, or toxic metabolites [6] within the central nervous system (CNS), which is composed of macroglia, microglia, neurons, and astrocytes. Neuroinflammation is considered to be a pathological mediator in a variety of neurodegenerative diseases [7]. The blood-brain barrier (BBB), a highly specialized structure made of endothelium and astrocytes, was previously considered to separate the CNS from the peripheral immune cells [4,8]. However, after injury and the prolonged release of inflammatory factors such as chemokines, the BBB is not only permeable to peripheral inflammation-induced proinflammatory mediators, but also allows leukocytes to migrate into the brain, which can induce pathogenesis in the CNS [9,10]. Dysfunction of the BBB facilitates neuroinflammation, giving rise to synaptic disruption, neuronal death, and aggravation of various brain-related diseases [2,11,12], which in turn aggravates chronic degenerative diseases including Alzheimer disease (AD), Parkinson disease (PD), amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS) [13-16]. In addition, these neurodegenerative diseases can also occur as a result of the activation of microglia [17] and pathways involving nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [18], pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin 1 beta (IL-1β) [4], mitochondrial dysfunction, and reactive oxygen species (ROS) [1,19].
Exercise has a beneficial impact on the whole body. It can improve cognitive function and brain health [20-23]. In response to exercise-related stimuli and mild injuries, the body activates the endogenous protective and recovery systems by altering gene expression and producing numerous factors involved in trophic effects, energy metabolism, antioxidation, and particularly, anti-inflammation [24-26]. These factors improve brain function and mitigate brain disorders by activating neuroplasticity, enhancing metabolic efficiency, and boosting antioxidative capacity [27,28]. Additionally, exercise maintains homeostasis of the brain and prevents brain pathology by modulating the activation of glia, pro-inflammatory cytokines, and neuroinflammation, thereby preventing neurodegenerative diseases such as AD, PD, ALS, and MS [24,29-31]. To determine the effects of exercise on neurodegenerative diseases, the type, intensity, frequency, and duration of exercise should be considered. Nonetheless, it is clear that exercise plays a protective role in neurodegenerative diseases by increasing levels of anti-inflammatory molecules and reducing those of pro-inflammatory molecules.
With this background, this review highlights the causes and consequences of neuroinflammation by focusing on (1) biological mechanisms that involve microglia, NF-κB, pro-inflammatory cytokines, and mitochondrial dysfunction in neurodegenerative diseases and (2) the effects of physical exercise on inflammation-related neurodegenerative diseases.
NEUROINFLAMMATION
Neuroinflammation Pathways
Microglia
Physiologically, microglia, which are the resident macrophages in the CNS, play a vital role in organism protection and tissue repair in the CNS [32]. Microglia scavenge plaques, unnecessary or disrupted synapses and neurons, and infectious agents in the CNS [33]. However, microglia that are activated by pathogens, abnormal stimulation, tissue damage, neurotoxins, injury, or infection are pivotal mediators in neuroinflammatory responses and neurodegenerative diseases [17]. More specifically, following the activation of microglia, they cause neuronal disruption and cell death by releasing proteins such as inducible nitric oxide synthase (iNOS), pro-inflammatory cytokines, including IL-1β, TNF-α, and cyclooxygenase-1 and 2 (COX-1, COX-2), ROS, and potential neurotoxic compounds, all of which induce neuroinflammation [34]. These proteins may attack healthy neurons, either by releasing pro-apoptotic factors or by phagocytosis [34].
NF-κB
NF-κB, located in almost all eukaryotic cells, is a protein complex that regulates DNA transcription, cytokine production, and cell survival [35]. NF-κB modulates multiple processes such as inflammation, immunity, apoptosis, cell survival, and the development of cancer; furthermore, it also controls the secretion of immune and inflammatory response genes [36]. NF-κB activation in glia plays a crucial role in the process of inflammation by causing neurodegeneration [37]. In particular, lipopolysaccharide (LPS), also known as endotoxin, causes systemic inflammatory response syndrome via toll-like receptor (TLR) signaling. LPS activates several signals, including phosphoinositide 3-kinase/protein kinase, mitogen-activated protein kinase, and mammalian target of rapamycin, which ultimately result in NF-κB activation [1]. Activated NF-κB then instigates the production of pro-neuroinflammatory mediators such as pro-inflammatory cytokines, inducible enzymes and chemokines, iNOS, and COX-2 [38].
Pro-inflammatory cytokines
Cytokines play a role in cell proliferation, survival, and death, and contribute to increased levels of leukocytes in the brain [39]. Even though physiological levels of cytokines, such as TNF-α and IL-1β, are important for synaptic plasticity during consolidation and memory formation, excessive cytokine levels are harmful [12,40]. As stated earlier, microglia induce TNF-α signaling, leading to inflammation and apoptosis [1]. For example, excessive apoptosis of hippocampal neurons is associated with high TNF-α level, suggesting that elevated concentrations of TNF-α can be an early indicator of apoptosis [4,41]. Similar to TNF-α, IL-1β has also been implicated in neuronal disruption, which is observed before neuronal death. It induces BBB breakdown, upregulates adhesion molecule expression, and promotes the spread of toxic substances such as nitric oxide [42]. Consequently, TNF-α and IL-1β play a pivotal role in acute neuroinflammatory conditions such as ischemia, stroke, and brain injury, and in chronic neurodegenerative diseases such as AD and PD [43].
Mitochondrial dysfunction and ROS
Mitochondria are organelles of eukaryotic cells that contribute to bioenergetic metabolism and regulate cellular homeostasis. They are involved in the generation of ATP using electron transport and oxidative phosphorylation, the initiation and execution of mitochondria-mediated apoptosis, and the production of ROS [44]. Mitochondria are a vital source of ROS, which are produced from the electron transport chain. ROS can have a toxic impact on biological macromolecules and can activate several genes that initiate inflammatory signaling cascades [1]. Although physiological levels of ROS regulate cell metabolism, excessive levels of ROS cause mitochondrial dysfunction and tissue injury through oxidative damage, leading to neuroinflammation [1]. Specifically, mitochondrial dysfunction in microglia results in excessive production of ROS. This promotes redox imbalance and regulates pro-inflammatory gene transcription and the expression of cytokines, such as IL-6, IL-1β, monocyte chemotactic protein-1, and TNF-α, by encouraging the expression of oxidative stress-modified mitochondrial DNA and polynucleotides, which causes inflammatory signaling in astrocytes [1,45].
Neuroinflammatory Disease
Alzheimer disease
It is well known that neuroinflammation plays a vital role in the development and progression of AD, which is a fatal neurodegenerative disorder that affects more than 15 million people worldwide [46]. Extracellular and intracellular protein aggregation contributes to the development of AD. Specifically, AD is characterized by a sequence of major pathogenic events and the presence of amyloid-β peptide (Aβ) plaques and neurofibrillary tangles (NFTs), which result in the formation of the microtubuleassociated protein tau, and the onset of synaptic and neuronal dysfunction and loss [47]. The accumulation of Aβ plaques, tau protein, and NFTs in the brain induces neuroinflammation and plays a pivotal role in regulating the pathogenesis of AD [48]. This causes neuronal dysfunction and increased expression of inflammatory mediators around Aβ plaques and NFTs [49]. In the early stages of AD, microglia are activated, leading to the production and secretion of neurotoxic cytokines such as TNF-α and IL-1β, the generation of ROS, the inhibition of neuroprotective effects, and mitochondrial dysfunction [50,51].
Parkinson disease
PD is a common and complex neurological disorder. It is a neurodegenerative disease involving the early prominent death of dopaminergic neurons in the substantia nigra pars compacta [52]. In addition, PD is also associated with numerous nonmotor symptoms, some of which precede motor dysfunction by more than a decade [53]. Even though the etiology of PD is unknown, according to some studies, PD is induced by inflammatory reactions [3], oxidative stress [4], mitochondrial dysfunction [5], proteotoxic stress [6], and kinase dysfunction [7], resulting in movement disorders like bradykinesia, which is responsible for postural instability and resting tremor [54]. These physical disorders result from the accumulation of α-synuclein protein, which forms insoluble Lewy bodies, and the selective loss of dopaminergic neurons in the substantia nigra pars compacta region of the brain, which is responsible for limiting movements [54-56]. The α-synuclein clusters and neuronal necrosis eventually activate the microglia. These microglia produce ROS, cytokines such as TNF-α, and chemokines [57]. Postmortem studies of the brains of PD patients have shown significant astrocyte activation and increased levels of various cytokines and microglia [58-60]. In addition, the increase in the levels of TNF-α and TNF-α receptors is an essential mediator of PD. This increase triggers the onset of extrinsic neuronal apoptosis, which is one of the key factors that induces PD [61].
Amyotrophic lateral sclerosis
ALS, also called Lou Gehrig’s disease, is the most common chronic motor neuron disease. It is characterized by selective motor neuron loss, weakness, and atrophy [62]. ALS is characterized by muscle stiffness and twitching, which insidiously worsen due to decreased muscle size. Although the cause of ALS is still unknown, potential evidence suggests that the innate immune system may be a focal contributor that promotes the activation of macrophages/microglia [63], which leads to the production of pro-inflammatory neurotoxic cytokines such as IL‐1β, thereby promoting the death of motor neurons [64]. Protein aggregates play a central role in superoxide dismutase 1 (SOD1)‐mediated ALS pathogenesis [65] and wild-type SOD1 aggregates have also been recently linked to sporadic ALS pathology [66]. Furthermore, ALS protein aggregates are robust immune response mediators in microglia [64]. The NOD-like receptor and the pyrin domain containing receptor 3 (NLRP3) inflammasome, an intracellular signaling complex, are a key factor in the innate immune system. Expression of the NLRP3 inflammasome is promoted by the aggregation of proteins and has been associated with neurodegenerative diseases [67]. Activation of the inflammasome requires a priming signal for the upregulation of NLRP3 and cytokine precursors, such as pro‐IL‐1β and pro‐IL‐18, followed by an activation step, which contributes to the recruitment of the inflammasome adapter, apoptosis‐associated speck‐like protein containing a caspase recruitment domain (ASC), the activation of caspase‐1 protease, and the cleavage and release of IL‐1β and IL‐18.
Multiple sclerosis
MS is a chronic autoimmune disease caused by CNS demyelination and inflammation, leading to damage of axons and myelin sheaths [4]. Although there might be a relationship between the development of the disease and systemic inflammation, there is little evidence supporting a relationship between inflammatory stimuli and the disrupted axons and myelin-producing cells [4]. Activation of the innate immune response, which involves the activation of microglia and macrophages, is responsible for the damaged axons observed during MS [68]. Systemic inflammation might contribute to the disruption of myelinated cells in MS. This explains the increased possibility of relapse following infection. Inflammation in MS results from the adaptive immune response, which involves T helper cells (Th1 and Th17) [69] and B cells [70]. Pro-inflammatory molecules generated by these glial cells and lymphocytes contribute to the initiation of MS [71].
EFFECTS OF EXERCISE ON NEUROINFLAMMATION
Exercise in AD
Several studies have demonstrated that exercise is a positive regulator of AD (Table 1). For example, Kim et al. [72] found that treadmill running decreased the Aβ plaque burden, neuro-inflammation, and mitochondrial dysfunction, suggesting that exercise enhanced the cognitive performance of 3xTg-AD mice. Similarly, Kim et al. [73] showed that 20 weeks of treadmill running ameliorated neuroinflammation and apoptotic neuronal cell death in high-fat-diet (HFD)-induced 3xTg-AD mice. These results suggest that treadmill running protects against AD pathology and cognitive deficiency in HFD-induced 3xTg-AD mice. In another study, wheel running was reported to increase microglia activation [74], attenuate microglia cytokine production, and protect against the negative effects of immune system activation [75]. Tapia-Rojas et al. [76] showed that voluntary running decreased neuronal loss, Aβ burden, and spatial memory loss, and increased neurogenesis in an AD animal model. Rodriguez et al. [77] reported that voluntary wheel running affected microglial density and activation-associated changes in microglial morphology. Zhang et al. [78] suggested that treadmill exercise significantly inhibited neuroinflammation by reducing the expression of pro-inflammatory factors and increasing the expression of anti-inflammatory factors. Exercise also attenuated oxidative stress induced by methane dicarboxylic aldehyde and dramatically elevated SOD and Mn-SOD activity. Therefore, treadmill exercise is a positive regulator of neuroinflammation and oxidative stress in AD. Sixteen weeks of treadmill running decreased the level of β-amyloid precursor protein (β-APP or Aβ peptide) in transgenic (TgCRND8) mice with AD phenotypes [79]. Treadmill running ameliorated ROS generation and mtDNA oxidative damage, increased the activity of mitochondrial antioxidant enzymes, and prevented mitochondrial dysfunction in APP/PS1 transgenic mice with an AD phenotype [80].

The effects of exercise on Alzheimer disease (AD)
Stress deterioration on the hippocampal function, leading to short-term memory problems has been shown, also, to impair lower urinary tract functions [81]. Whereby, Heo et al. [82] investigated, not only CNS effects, but concomitant renal injuries associated with short-term memory disturbance. Treadmill exercise ameliorated short-term memory impairment, suppressed AChE expression and enhanced angiogenesis in mice with scopolamine-induced amnesia.
Exercise in PD
PD is a neurodegenerative disease characterized by the death of dopaminergic neurons, leading to decreased dopamine transmission and changes in motor and cognitive function [83,84]. Exercise may be one of the most promising therapeutic approaches because it inhibits the factors that promote neurodegenerative diseases and increases the levels of neurotrophic factors, resulting in a healthy CNS in the elderly population [85,86]. Real et al. [87] showed that 4 weeks of treadmill running reduced neuroinflammatory processes, thereby decreasing the risk of PD development, the activation of astrocytes and microglia, and the oxidative stress response. Tuon et al. [88] also reported that treadmill running modulated α-synuclein activity, brain-derived neurotrophic factor (BDNF), and sarcoplasmic reticulum Ca2+ ATPase (SERCA) II levels for 8 weeks in the striatum of a PD animal model. These results demonstrated that exercise protected the dopamine system in a 6-hydroxydopamine-induced PD animal model. Koo et al. [89] reported that 8 weeks of treadmill exercise inhibited TLR2 expression, microglial activation, neuroinflammation, ROS production, and apoptosis, and suppressed the expression of the nicotinamide adenine dinucleotide phosphate oxidase subunit, NF-κB, TNF-α, and IL-1β in a preclinical model of PD. The authors showed that treadmill exercise is a nonpharmacological tool for managing neurodegeneration in PD. Human studies confirmed the beneficial effect of exercise by demonstrating that intensive exercise resulted in a 16% increase in serum BDNF levels and improvements in scores on the Unified Parkinson’s Disease Rating Scale, which evaluates the benefits of therapeutic interventions [90]. In another study, moderate exercise for 8 weeks increased the level of serum BDNF, decreased the level of serum vascular cell adhesion molecule by 25%, and reduced the level of serum TNF-α in PD patient [91]. Bloomer et al. [92] reported that 8 weeks of resistance exercise decreased the oxidative stress caused by malondialdehyde and hydrogen peroxide. The study also confirmed an increase in the levels of SOD (9%) and glutathione peroxidase (15%), but these changes were not significant in PD patients. These central effects seem to propagate across to peripheral systems, as well. In a study applying treadmill exercise on PD rat models, Lee et al. [93] showed improvement of cerebellar functions by inhibiting Purkinje cell apoptosis. A summary of the effects of exercise on PD is shown in Table 2.

The effects of exercise on Parkinson disease (PD)
Exercise in ALS
Exercise is considered to be a key regulator in ALS, a chronic motor neuron disease. Exercise prevents ALS by regulating neuroinflammation. The effects of exercise in ALS are summarized in Table 3. Just-Borràs et al. [62] showed that moderate exercise, including running and swimming, maintained the BDNF/TrkB signaling pathway and downstream signaling for 45 days in an ALS animal model. These results are encouraging, since they show improvements even when therapy is started after the onset of the disease. Flis et al. [94] evaluated the effects of swimming exercise for 15 weeks on oxidative stress and mitochondrial function in an ALS animal model. They observed that mitochondrial function was maintained and oxidative stress was lowered, and that there was an exercise-induced deceleration in ALS development. Recently, Flis et al. [95] also reported that there were no significant changes in malondialdehyde, COX activity, and mitochondria oxygen consumption in an ALS mouse model after 15 weeks. In another study, Kassa et al. [96] reported treadmill running-induced increases in microglia activation and motor neuron counts in ALS mice. This suggests that exercise can ameliorate ALS symptoms and progression [97]. Taken together, exercise of various types and intensities can influence various aspects of ALS.

The effects of exercise on amyotrophic lateral sclerosis (ALS)
Exercise in MS
There is compelling evidence for the beneficial effects of exercise in MS. Accumulating evidence supports the role of exercise in neuroinflammation in MS (Table 4). For example, Kierkegaard et al. [98] reported that resistance exercise decreased plasma TNF-α level for 12 weeks in MS patients. Another study showed that resistance training significantly reduced serum levels of cytokines, including IL-4, IL-10, C-reactive protein, and interferon-gamma (IFN-γ) in MS patients [99]. In another study, Donia et al. [100] suggested that 1 hour of moderate aerobic exercise (60% of peak oxygen uptake) decreased plasma TNF-α levels in MS patients. Interestingly, combined aerobic training and Pilates training improved BDNF and physical performance, suggesting that combining forms of exercise might yield a beneficial effect in MS patients [101]. Castellano et al. [102] demonstrated that acute and long-term cycle exercise, which involved 60% of peak O2 uptake for 3 days per week over the course of 8 weeks reduced plasma TNF-α, IL-6, and IFN-γ levels in MS patients. Further investigations will help to elucidate the effects of exercise on neuroinflammation in MS.
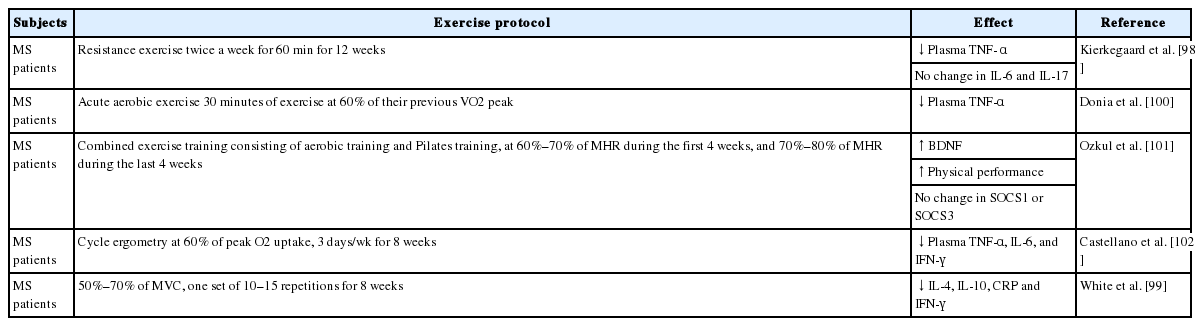
The effects of exercise on multiple sclerosis (MS)
CONCLUSIONS
In this article, we highlighted neuroinflammation-related diseases, such as AD, PD, ALS, and MS. These diseases are associated with the activation of microglia, NF-κB, pro-inflammatory cytokines, mitochondrial dysfunction, and ROS. Even though further research is needed to confirm our findings, exercise might mitigate neuroinflammation in AD, PD, ALS, and MS. In order to elucidate the cellular and/or molecular mechanisms that underlie the role of exercise in attenuating the activation of microglia, NF-κB, pro-inflammatory cytokines, mitochondrial dysfunction, and ROS in the brain, further clinical and preclinical studies should be conducted.
Notes
Fund/Grant Support
This work was supported by the Ministry of Education of the Republic of Korea and the National Research Foundation of Korea (2016R1A2B4014240, 2018R1A2A3074577, 2019S1A5C2A03082727).
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION STATEMENT
·Full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis: DYS, HBK
·Study concept and design: DYS, HBK
·Acquisition of data: JWH, JRK
·Analysis and interpretation of data: JWH, JRK
·Drafting of the manuscript: DYS, HBK
·Critical revision of the manuscript for important intellectual content: HBK
·Administrative, technical, or material support: HBK
·Study supervision: HBK