INTRODUCTION
Heart failure (HF) is a multicomponent disease wherein the heart is unable to provide sufficient oxygen-rich blood for the metabolic needs of the surrounding tissues. Recent technological and pharmacological trends have helped in decreasing mortalities due to HF, however, the disease still poses danger to about 5.8 million people in the United States alone, in addition to 23 million more people worldwide [1]. The Framingham Heart Study approximates 1-month to 1-year mortality at around 20%–30%, and 5-year mortality between 45%–60% [2]. It should be kept in mind that HF is a rather complex syndrome, whose progression depends on factors such as physiological conditions, preexisting cardiovascular diseases like coronary heart disease (CHD), high blood pressure, and diabetes; and occurs together with diseases that further aggravate the condition, such as diabetes and hypertension [3]. CHD alone, specifically coronary artery disease, was responsible for 65% of patients afflicted with HF [4]. It has also been suggested that neurological dysfunction further contributes to the progression of CHD. CHD has been directly linked with bladder pain syndrome/interstitial cystitis, a recurring discomfort around the pelvic and bladder area, wherein it was concluded that endothelial dysfunction might be a pathogenic factor common between the two conditions [5]. The pathomechanism remains unclear; however, the progression and coexistence of these diseases, which lead to HF, might be linked with mitochondrial irregularities.
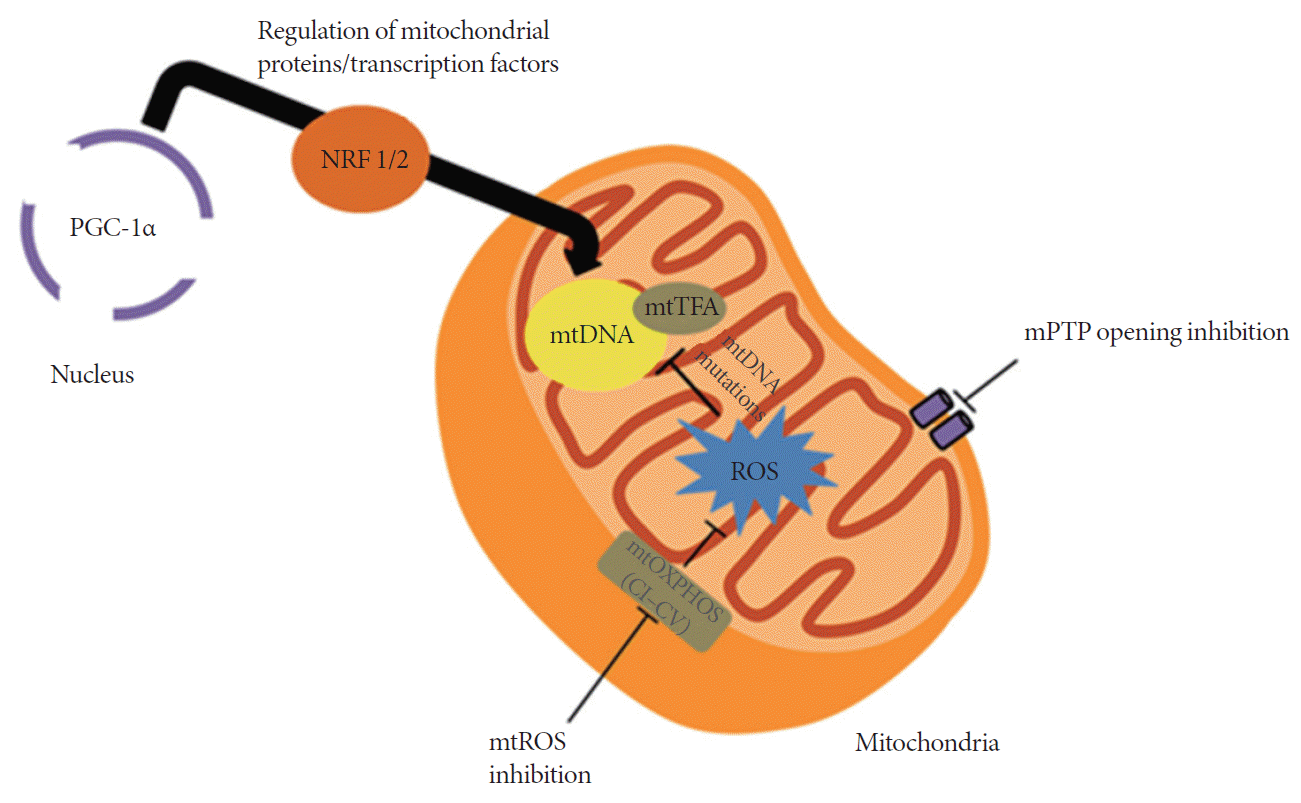
The pervasiveness of HF is due, in part, to factors such as irregularities in signal transduction pathways and mitochondrial deterioration [6]. The role of mitochondria in the production of adenosine triphosphate (ATP) and in the regulation of cell death makes it vital for cell survival. Data published in recent years suggest that mitochondria might be pivotal in HF development [7]. The levels of oxidative stress brought about by reactive oxygen species (ROS) as well as regulation of opening/closing of the mitochondrial permeability transition pore (mPTP) also contribute to HF development. Previous studies have implicated altered mitochondrial biogenesis as one of the causal mechanisms of oxidative phosphorylation (OXPHOS) dysfunction in cardiac remodeling [8]. It will be important to focus on restoring contractile function in failing hearts by targeting the mitochondria. This review outlines some common and recent modes of mitochondrial targeting for HF, and provides insights on future directions for HF treatment (Fig. 1).
MITOCHONDRIAL TARGETS
Mitochondrial Biogenesis
Primary alterations in mitochondrial biogenesis may be associated with the progression of cardiac pathologies. Mitochondrial DNA (mtDNA) copy number and mitochondrial content are significantly reduced in both human and rat models of failing myocardium, which can be attributed to downregulation of the mitochondrial biogenesis signaling pathways [9-12]. It is suggested that a disturbance in mitochondrial biogenesis occurs at early onset of HF, which is cardioprotective upon reversal. Recently, peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) has been studied for its central role in the regulation of transcription factors in the mitochondria, such as nuclear respiratory factor 1/2 (NRF 1/2) and mitochondrial transcription factor A (mtTFA). PGC-1α is a protein encoded in the nucleus and is activated during periods of high energy demand, such as those during increased cardiac workload, physical training or exercise, or starvation. PGC-1α is responsible for the activation of mitochondrial proliferation through its intercommunication with different transcription factors. PGC-1α was evidently decreased in various HF experimental models, and this affected essential, related transcription factors, as evidenced by decreased NRF 1/2 and mtTFA expression [13,14]. Targeted mtTFA disruption in cardiac tissue affected electron transport chain (ETC) capacity, eventually resulting in spontaneous cardiomyopathy and HF [15,16].
Increased mtDNA and mitochondrial proliferation with myofibril displacement is associated with primary mitochondrial defects and cardiomyopathy, and commonly related with increased mitochondrial biogenesis-related gene expression [17]. It was previously observed that mitochondrial proliferation was higher in cardiomyopathic murine models, and associated with the removal of adenine nucleotide translocase 1 (ANT1) [18], manganese superoxide dismutase (Mn-SOD) [19], and mtTFA [20]. Furthermore, there was increased mitochondrial mass in mtTFA-knocked out mice; in addition to decreased levels of mtDNA and cytochrome c oxidase subunit I, peroxisome proliferator-activated receptors α (PPARα), and PPARα-dependent transcripts; and dysfunctional ETC [21]. The significant decrease in ATP revealed that increase in mitochondrial number is not enough to compensate for damage in the mitochondria. These examples demonstrate the importance of improved mitochondrial biogenesis and mtDNA copy number in cardioprotection against HF.
Recent studies have focused on targeting the mitochondria and improving mitochondrial biogenesis through various methods, including those involving pharmacological agents. Currently, there are no pharmacologic agents that specifically target mitochondrial biogenesis in HF. However, stimulation of mitochondrial biogenesis, leading to its beneficial effects, is accelerated by targeting other components such as endothelial nitric oxide synthase, adenosine monophosphate-activated kinase, and other mitochondrial biogenesis pathways, which all hold promise in addressing the disease [22,23]. A summary of some common and recent experimental and clinical interventions are presented in Table 1. One of the earlier drugs to be used, an angiotensin-converting enzyme (ACE) inhibitor named captopril (now marketed as Capoten), increased mitochondrial content in canine hearts after coronary ligation [24]. This indicates that the favorable effects may, in part, be related to mitochondrial invigoration. Current studies have examined the effects of various ACE inhibitors in human patients, including that of captopril. Although captopril is widely used, patients treated with the drug presented higher incidence of coughing, and it was suggested that enalapril is more potent, owing to its effect on ejection fraction, stroke volume, and mean arterial pressure [25].
Mitochondrial Oxidative Stress
Mitochondrial oxidative stress, caused by either ROS overproduction or decreased endogenous antioxidant defenses, has been implicated in the structural and functional alterations during myocardial failure [39]. Sources of ROS in the failing heart include mitochondrial ETC, nicotinamide adenine dinucleotide phosphate oxidases, nitric oxide synthases, and xanthine oxidase [40]. The change in oxidative stress levels may contribute to changes in the abundance and copy number of mitochondria, and in the integrity of mtDNA in human cells during pathological conditions [41]. ROS may act within a threshold to generate stress responses through various cellular processes, such as modifications in specific nuclear genes, in order to maintain energy metabolism and production [42]. Decreased mitochondrial biogenesis occurs during insignificant increases in oxidative stress, in addition to increased mitochondrial DNA mutations, which eventually lead to impaired OXPHOS and cardiomyopathy. On the other hand, induction of PGC-1α, mitochondrial biogenesis, and enzymes related to metabolism were observed during marked elevation in oxidative stress [43].
The complex role of ROS in the progression of HF remains to be clarified. A suggested mechanism involves impairment of cellular and mitochondrial structures, such as excitation-contraction coupling proteins, which affect the mechanical function of the heart [44]. Considering that majority of the ROS originates in the mitochondria during HF, it is not surprising that these organelles are the most susceptible to oxidative damage. In fact, mtDNA is more prone to ROS damage since it lacks histones, which can serve as protection from ROS attack. This renders the mtDNA to a less efficient DNA repair system, which results in mtDNA gene mutations. Furthermore, decrease in PGC-1α in affected hearts aggravates oxidative stress and mitochondrial damage, owing to the protein’s role in maintaining mitochondrial antioxidant defenses [43]. ROS also regulates signaling cascades such as those involving protein kinase C (PKC), mitogen-activated protein kinase (MAPK), Jun N-terminal kinase, and Ras—all implicated in hypertrophy [45]. Further, ROS facilitates extracellular matrix remodeling by promoting matrix metalloproteinases either directly via posttranslational modifications (PTMs) or indirectly via nuclear factor κB (NFκB) pathway [46]. The aforementioned roles of ROS highlight its importance and novelty as a therapeutic target for treating HF. As mentioned in the previous section, therapies involving ACE inhibitors and angiotensin II receptor blockers have been used experimentally, owing to the agents’ antioxidant properties. However, it is unclear whether these agents target mitochondrial ROS directly or indirectly [47,48].
Numerous studies have focused on oxidative-stress targets or energetics in HF models. Large randomized trials of antioxidant vitamins such as vitamin E have so far been futile, which might be due to the nonspecific nature of the vitamins, possibly inhibiting both the beneficial and negative effects of ROS. Nevertheless, it has been shown that mitochondrial targeting of ROS-scavenging molecules is protective. Modulation of substrate utilization with drugs such as perhexiline to increase myocardial energy production has only been performed in small-scale clinical studies [49]. A two-pronged approach of addressing oxidative stress and mitochondrial dysfunction together has been deemed a more efficient approach. The use of antioxidants such as triphenylphosphonium conjugation (MitoQ) and novel Mn-SOD, or catalase analogues could be considered in future HF studies.
Mitochondrial Permeability Transition Pore
Decreased energy metabolism due to dysfunction in mitochondria is further aggravated by ROS generation, which, when taken together with Ca2+ mishandling, favors mPTP opening. mPTP is a nonselective pore in the inner mitochondrial membrane, activated under high Ca2+ and ROS levels, resulting in mitochondrial dysfunction and inhibition of OXPHOS [50,51]. It was earlier thought that the inner-membrane ANT [52], outer-membrane voltage-dependent anion channel [53], and inner phosphate carrier [54] all play essential roles in mPTP induction. However, recent studies have pointed out that these components only play a supporting, regulatory role, while more compelling evidence points at the importance of matrix protein cyclophilin D (CyP-D) and mitochondrial ATPase in mPTP structure and regulation [55]. Determining the structure of mPTP would aid the development of better methods to inhibit mPTP opening, helping in the maintenance of inner-membrane integrity and preservation of ATP production, which would prevent cell death during the onset of HF.
mPTP is widely studied in cardiac pathology, including ischemia/reperfusion (I/R) and HF. It was earlier observed that inhibition of mPTP opening by cyclosporine A (CsA) offers possible cardioprotection. Subsequent research in a myocyte model of hypoxia/reoxygenation found that CsA is a potent inhibitor of mPTP pore in isolated mitochondria. Earlier studies involving CsA also revealed cardioprotection from reoxygenation injury in isolated cardiac myocytes [56]. This was supported by another study where CsA exhibited cardioprotection from reperfusion injury in the Langendorff perfused heart [57]. Cardioprotective effects of CsA were also observed in CyP-D-knocked out mice, exhibiting a significant reduction in infarct size in an I/R model [58]. However, owing to its affinity to cytosolic cyclophilin-A, CsA poses a problem regarding cardioprotection, wherein it inhibits calcineurin, which directly affects heart function [59] and is reported to possess immunosuppressive activity [60]. In order to address such problems exhibited by CsA, analogues have been developed that are inactive against calcineurin, but still bind strongly to CyP-D. The cardioprotective properties of these analogues are similar to that of CsA, and are potent even when administered only during early reperfusion [61-64].
Several other indirect inhibition methods for mPTP have been suggested, mostly focused on reduction of mitochondrial build-up of pore-formation inducers such as ROS and Ca2+. Considering that oxidative stress is a robust mPTP-opening activator, clinical use of ROS scavengers might be effective [65]. Propofol, which is used during cardiac surgeries, inhibited mPTP opening in an I/R model of isolated rat hearts, exhibiting significant post-ischemic recovery of heart function. This cardioprotective effect could be attributed to propofol’s role in inhibiting the formation of ROS–cardiolipin complexes in the respiratory chain by maintaining the integrity of cardiolipin under ROS attack [66]. In another case, 3-Methyl-1-phenyl-2-pyrazolin-5-one (MCI-186) significantly decreased myocardial infarction size and inhibited mPTP opening in an I/R rat model. This effect could be attributed to the action of MCI-186 in inhibiting cellular Ca2+ overload brought about by oxidative stress, in addition to the inhibitory effect on mitochondrial swelling and cytochrome c release [67].
Ca2+ channel blockers have been well studied in cardiac disease models, but the role of mPTP in relation to these blockers remains unclear. Ruthenium 360 is a specific Ca2+ uniport inhibitor, which reduces mitochondrial Ca2+ by blocking mPTP while not affecting Ca2+ movement. This was associated with cardiac function recovery after ischemia [68]. Hearts administered with Na+/H+ exchanger isoform 1 (NHE-1) inhibitors showed decreased muscle cell injury as evidenced by low lactate dehydrogenase release and improved cardiac function during reperfusion [69]. The NHE-1 inhibitor cariporide was also able to mitigate oxidative stress-induced mitochondrial Ca2+ overload and mitochondrial membrane potential (ΔΨm) loss in neonatal cardiomyocytes [70]. Thus, there is a need to develop drugs that are more potent in targeting other aspects of mPTP, without disrupting normal mitochondrial function.
FUTURE DIRECTION AND CONCLUSION
Mitochondria are deemed pivotal in both normal and pathological functioning of the heart, and are implicated as either the primary cause or an aid in the progression of HF. There are many other factors that are yet to be considered in order to come up with more successful methods of dealing with HF. Pharmacological intervention may be used in patients at the risk of HF, especially when addressing novel cardiac targets such as mitochondrial PTM [71]. Briefly, PTMs are conformational changes that occur in a protein after the translational process, in response to external stimuli like ROS or aberrant signaling, and these PTMs significantly affect cellular function [7].
Taken together, maintaining mitochondrial biogenesis against cardiac injury and decreasing mitochondrial ROS are two promising mechanisms that may lead to effective treatments for HF. In addition, results from experiments show that mPTP also holds promise as a target in HF treatment; these experiments should be followed up.